Лентигиноз при синдроме Пейтца-Егерса
Добавил пользователь Алексей Ф. Обновлено: 07.01.2026
Появление пигментных пятен кофейного цвета в области губ и слизистой оболочки рта может свидетельствовать о наличии редкого онкологического синдрома.
На консультацию к врачу-генетику Центра персонализированной медицины МКНЦ обратилась пациентка М. в возрасте 35 лет с диагнозом множественный полипоз толстой кишки, рак толстой кишки. Из активных жалоб отмечалась общая слабость, недомогание, периодические боли в животе и нарушение стула на протяжении длительного времени.
За несколько месяцев до этого пациентка обнаружила кровь в стуле, после чего обратилась за помощью в поликлинику по месту жительства, где при колоноскопии выявлено более 50 полипов толстой кишки и заподозрено злокачественное новообразование. Пациентка сообщила, что ее мать умерла от рака толстой кишки в возрасте 40 лет. При осмотре обращала на себя внимание светлая фарфоровая кожа с множеством веснушчатых пигментных пятен, равномерно расположенных по всему телу, рыжий цвет волос и светлый оттенок глаз. Однако скопление пигментаций в области губ усиливалось, они переходили на слизистую оболочку полости рта и щек, где размеры пятен достигали 5 мм в диаметре. Пациентка пришла на прием с родной сестрой, у которой подобных особенностей не наблюдалось.
Учитывая данные осмотра, отягощенный семейный анамнез раком толстой кишки и выявление полипов при колоноскопии у пациентки заподозрен синдром Пейтца-Егерса, в связи с чем наши специалисты рекомендовали проведение молекулярно-генетической диагностики для исключения наследственной природы заболевания и разработали индивидуальный план наблюдения.
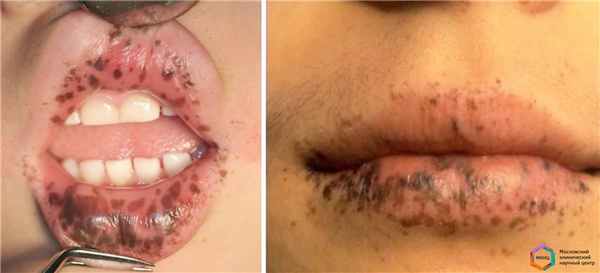
Синдром Пейтца-Егерса – это редкий наследственный онкологический синдром, который развивается в результате нарушений в строении гена STK11. Классически этот синдром проявляется скоплением множества пигментных пятен кофейного цвета размерами до 5-7 мм. Преимущественно они располагаются на губах, слизистой оболочке рта, в некоторых случаях на кончиках пальце или стопах и наблюдаются с детства. Второй особенностью данного синдрома является наличие множества полипов (доброкачественных новообразований) желудочно-кишечного тракта, которые способны к злокачественной трансформации. Наиболее часто полипы выявляются в тонкой кишке, но также могут встречаться в желудке и толстой кишке. На протяжении жизни они могут изъязвляться, кровоточить, вызывать кишечную инвагинацию и непроходимость, что приводит к установлению диагноза в возрасте до 20 лет у 50% больных.
Известно, что у пациентов с синдромом Пейтца-Егерса риски возникновения злокачественных новообразований в различных органах в несколько раз выше, чем без мутации в гене STK11. Пациенты с данным синдромом нуждаются в составлении индивидуального плана периодического наблюдения, в который обязательно входит колоноскопия, маммография, эзофагогастродуоденоскопия, эндоскопическое исследование тонкой кишки, гинекологическое обследование у женщин. В случае обнаружения подозрительных полипов специалисты рекомендуют их эндоскопическое иссечение - полипэктомия.
Учитывая, что риски наследования данной особенности составляют 50%, при обнаружении мутации у пациента рекомендуется консультация врача-генетика и проведение молекулярно-генетической диагностики у его родственников для своевременного оказания специализированной медицинской помощи.
Синдром Пейтца-Егерса: что стало известно за 125 лет изучения? (обзор литературы)
Синдром Пейтца-Егерса (СПЕ) является крайне редким аутосомно-доминантным наследственным заболеванием, которое клинически характеризуется ростом гамартомных полипов в желудочно-кишечном тракте, слизисто-кожной пигментацией и повышенным риском возникновения злокачественных новообразований различной локализации. В большинстве случаев развитие СПЕ связано с наличием мутации в гене STK11, однако не у всех пациентов имеется данная мутация. В настоящем обзоре литературы представ- лены исторические аспекты появления данных о СПЕ, рассмотрены клинические проявления заболевания, актуальные методы диагностики, а также современные знания о генетических причинах развития СПЕ, риске возникновения злокачественных новообразований у пациентов с СПЕ, существующие рекомендации по скринингу и лечению пациентов с СПЕ. Однако наличие ряда нерешенных до настоящего времени вопросов в генетике, мониторинге и лечении свидетельствуют о необходимости дальнейших исследований.
Ключевые слова
Об авторах
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
Список литературы
1. Kopacova M, Tacheci I, Rejchrt S. et al. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15(43):5397–5408.
2. Цуканов А.С., Шелыгин Ю.А., Фролов С.А. и соавт. Семейный аденоматоз толстой кишки. Хирург. 2017;3:14–23.
3. Шелыгин Ю.А., Кашников В.Н., Фролов С.А. и соавт. Молекулярно-генетическое исследование наследственной пред- расположенности к разным формам полипоза толстой кишки. Колопроктология. 2013;1(43):9–14.
4. Giardiello F, Trimbath J. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4(4):408–415.
5. Schreibman I, Baker M, Amos C. et al. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005 Feb;100(2):476–90.
6. McGarrity T, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci. 2006;63(18):2135–2144.
7. Hutchinson J. Pigmentation of lips and mouth. Archives of Surgery. 1896;7:290–291.
8. Peutz J. Over een zeer merkwaardige, gecombineerde familiaire polyposis van de slijmliezen van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid-enslijmvliezen. Ned Maandschr Gen. 1921;10:134–146.
9. Jeghers H, McKusick V, Katz K. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:1031–1036.
10. Bruwer A, Bargen J, Kierland R. Surface pigmentation and generalized intestinal polyposis; (Peutze-Jeghers syndrome). Proc Staff Meet Mayo Clin. 1954;29:168–171.
11. Beggs A, Latchford A, Vasen H. et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986.
14. Hinds R, Philp C, Hyer W. et al. Complications of childhood Peutz- Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004;39(2):219–20.
15. Turpin A, Cattan S, Leclerc J. et al. Prédisposition héréditaire aux cancers digestifs, mammaires, gynécologiques et gonadiques : état des lieux du syndrome de Peutz-Jeghers [Hereditary predisposition to cancers of the digestive tract, breast, gynecological and gonadal: focus on the Peutz-Jeghers]. Bull Cancer. 2014;101(9):813–822.
16. Gao H, van Lier M, Poley J. et al. Endoscopic therapy of smallbowel polyps by double-balloon enteroscopy in patients with Peutz- Jeghers syndrome. Gastrointest Endosc. 2010;71(4):768–773.
17. Vogel T, Schumacher V, Saleh A. et al. Extraintestinal polyps in Peutz-Jeghers syndrome: presentation of four cases and review of the literature. Deutsche Peutz-Jeghers-Studiengruppe. Int J Colorectal Dis. 2000;15(2):118–123.
18. Van Lier M, Mathus-Vliegen E, Wagner A. et al. High cumulative risk of intussusceptions in patients with Peutz-Jeghers syndrome: time to update surveillance guidelines? Am J Gastroenterol. 2011;106(5):940–945.
19. Tse J, Wu S, Shinagare S. et al. Peutz-Jeghers syndrome: a critical look at colonic Peutz-Jeghers polyps. Mod Pathol. 2013;26(9):1235–1240.
20. Choi H, Park Y, Youk E. et al. Clinical characteristics of Peutz- Jeghers syndrome in Korean polyposis patients. Int J Colorectal Dis. 2000;15(1):35–38.
21. Kamilaris C, Faucz F, Voutetakis A. et al. Carney Complex. Exp Clin Endocrinol Diabetes. 2019;127(2–03):156–164.
22. Sarkozy A, Conti E, Digilio M. et al. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 2004;41(5):e68.
23. Aretz S, Stienen D, Uhlhaas S. et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513–519.
24. Ober W. Selected items from the history of pathology: Eugen Albrecht, MD (1872–1908): hamartoma and choristoma. Am J Pathol. 1978;91(3):606.
25. Jansen M, de Leng W, Baas A. et al. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut. 2006;55(1):1–5.
26. Аруин Л.И., Капуллер Л.Л., Исаков В.А. Морфологическая диагностика болезней желудка и кишечника. Москва: «Триада-Х». 1998; с. 441.
28. Hemminki A, Markie D. Tomlinson I. et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187.
29. Van Lier M, Wagner A, Mathus-Vliegen E. et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258–1265.
30. Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007 Dec 13;26(57):7825–32.
32. Jishage K, Nezu J, Kawase Y. et al. Role of LKB1, the causative gene of Peutz-Jegher’s syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99(13):8903–8908.
33. Tiainen M, Vaahtomeri K, Ylikorkala A, Mäkelä T. Growth arrest by the LKB1 tumor suppressor: induction of p21 (WAF1/CIP1). Hum Mol Genet. 2002;11(13):1497–1504.
34. Sfakianaki M, Papadaki C, Tzardi M. et al. Loss of LKB1 Protein Expression Correlates with Increased Risk of Recurrence and Death in Patients with Resected, Stage II or III Colon Cancer. Cancer Res Treat. 2019 Oct;51(4):1518–1526.
35. Schumacher V, Vogel T, Leube B. et al. STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet. 2005;42(5):428–435.
36. Chen C, Zhang X, Wang D. et al. Genetic Screening and Analysis of LKB1 Gene in Chinese Patients with Peutz-Jeghers Syndrome. Med Sci Monit. 2016 Oct 10;22:3628–3640.
37. Vélez A, Gaitan M, Marquez J. et al. Two novel LKB1 mutations in Colombian Peutz-Jeghers syndrome patients. Clin Genet. 2009;75(3):304–306.
38. Amos C, Keitheri-Cheteri M, Sabripour M. et al. Genotypephenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41(5):327–333.
39. Mehenni H, Resta N, Park J. et al. Cancer risks in LKB1 germ line mutation carriers. Gut. 2006;55(7):984–990.
40. Giardiello F, Brensinger J, Tersmette A. et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453.
41. Hearle N, Schumacher V, Menko F. et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215.
42. Daniell J, Plazzer J, Perera A, Macrae F. An exploration of genotype- phenotype link between Peutz-Jeghers syndrome and STK11: a review. Fam Cancer. 2018;17:421–427.
43. Giardiello F, Welsh S, Hamilton S. et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316(24):1511–1514.
44. Utsunomiya J, Gocho H, Miyanaga T. et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136(2):71–82.
45. Van Lier M, Westerman A., Wagner A. et al. High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome. Gut. 2011;60:141–147.
46. Resta N, Pierannunzio D, Lenato G. et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45:606–611.
47. McGarrity T, Amos C, Baker M. Peutz-Jeghers Syndrome. 2001 Feb 23 [updated 2016 Jul 14]. In: Adam M., Ardinger H., Pagon R., Wallace S. et al. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020.
48. Chen H, Jin X, Li B. et al. Cancer risk in patients with Peutz- Jeghers syndrome: A retrospective cohort study of 336 cases. Tumour Biol. 2017;39(6):1010428317705131.
49. Taheri D, Afshar-Moghadam N, Mahzoni P. et al. Cancer problem in Peutz-Jeghers syndrome. Adv Biomed Res. 2013;2:35. 50. Bosman F. The hamartoma–adenoma–carcinoma sequence. J Pathol. 1999;188:1–2.
51. Bouraoui S, Azouz H, Kechrid H, et al. PeutzeJeghers’ syndrome with malignant development in a hamartomatous polyp: report of one case and review of the literature (In French). Gastroenterol Clin Biol. 2008;32:250–254.
52. Hizawa K, Iida M, Matsumoto T. et al. Neoplastic transformation arising in Peutz-Jeghers polyposis. Dis Colon Rectum. 1993;36:953–957.
53. Latchford A, Phillps R. Gastointestinal polyps and cancer in Peutz- Jeghers syndrome: clinical aspects. Fam Cancer. 2011;10:455–61.
54. Шелыгин Ю.А., Поспехова Н.И., Шубин В.П. и соавт. Пилотное клинико-генетическое исследование российских пациентов с син- дромом Пейтца-Егерса. Вопросы онкологии. 2016;62(1):112–116.
55. Pilleul F, Penigaud M, Milot L. et al. Possible small-bowel neoplasms: contrast-enhanced and water-enhanced multidetector CT enteroclysis. Radiology. 2006 Dec;241(3):796–801.
56. Tomas C, Soyer P, Dohan A. et al. Update on imaging of Peutz-Jeghers syndrome. World J Gastroenterol. 2014 Aug 21;20(31):10864–75.
57. Caspari R, von Falkenhausen M, Krautmacher C. et al. Comparison of capsule endoscopy and magnetic resonance imaging for the detection of polyps of the small intestine in patients with familial adenomatous polyposis or with Peutz-Jeghers’ syndrome. Endoscopy. 2004;36:1054–1059.
58. Ross A, Dye C, Prachand V. Laparoscopic-assisted double-balloon enteroscopy for small-bowel polyp surveillance and treatment in patients with Peutz-Jeghers syndrome. Gastrointestinal Endoscopy. 2006;64:984–988.
59. Postgate A, Hyer W, Phillips R. et al. Feasibility of video capsule endoscopy in the management of children with Peutzejeghers syndrome: a blinded comparison with barium enterography for the detection of small bowel polyps. J Pediatr Gastroenterol Nutr. 2009;49:417–23.
60. Su X, Ge Y. Liang B.et al. Small intestinal tumors: diagnostic accuracy of enhanced multi-detector CT virtual endoscopy. Abdom Imaging. 2012 Jun;37(3):465–74.
61. Jee Hee Son, Bo Young Chung, Min Je Junget al. Cowden Disease: Case Report and Review of the Literature. Ann Dermatol. 2019;Jun;31(3):325–330.
62. Hearle N, Schumacher V, Menko F. et al. STK11 status and intussusception risk in Peutz-Jeghers syndrome. J Med Genet. 2006;43(8):e41.
63. Spigelman A, Thomson J, Phillips R. Towards decreasing the relaparotomy rate in the Peutz-Jeghers syndrome: the role of peroperative small bowel endoscopy. Br J Surg. 1990;77(3):301–302.
64. Amaro R., Diaz G., Schneider J.et al. Peutz-Jeghers syndrome managed with a complete intraoperative endoscopy and extensive polypectomy. Gastrointest Endosc. 2000;52(4):552–554.
65. Edwards D, Khosraviani K, Stafferton R, Phillips R. Long-term results of polyp clearance by intraoperative enteroscopy in the Peutz-Jeghers syndrome. Dis Colon Rectum. 2003;46(1):48–50.
66. Sakamoto H, Yamamoto H, Hayashi Y. et al. Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-balloon endoscopy. Gastrointest Endosc. 2011;74(2):328–333.
67. Robinson J, Lai C, Martin A. et al. Oral rapamycin reduces tumour burden and vascularization in LKB1+/– mice. J Pathol. 2009;219(1):35–40.
68. Kuwada S, Burt R. A rationale for mTOR inhibitors as chemoprevention agents in Peutz-Jeghers syndrome. Fam Cancer. 2011;10(3):469–472.
69. Ishida H, Tajima Y, Gonda T. et al. Update on our investigation of malignant tumors associated with Peutz-Jeghers syndrome in Japan. Surg Today. 2016 Nov;46(11):1231–42.
Cиндром Пейтца-Йегерса
Синдром Пейтца – Йегерса – заболевание с аутосомно-доминантным типом наследования, при котором наблюдается развитие множественных гамартомных полипов в желудке, тонкой и толстой кишке, а также характерных пигментированных участков на коже и слизистых.
Большинство (от 66 до 94%) случаев, по-видимому, вызваны мутацией зародышевой линии гена-супрессора опухоли STK11/LKB1 (серин/треонин-киназа 11). У больных существенно повышен риск рака желудочно-кишечного тракта и другой его локализации. Со стороны ЖКТ могут развиваться рак поджелудочной железы Рак поджелудочной железы Рак поджелудочной железы, в первую очередь протоковая аденокарцинома, составляет приблизительно 57 600 случаев и приводит к 47 050 летальных исходов в США ежегодно ( 1). Клинические проявления. Прочитайте дополнительные сведения , желудка Рак желудка Этиология рака желудка рассматривается как многофакторная, при этом показана важная роль Helicobacter pylori. Проявления болезни включают чувство раннего насыщения, нарушение проходимости. Прочитайте дополнительные сведения , тонкой кишки Опухоли тонкой кишки Опухоли тонкой кишки составляют 1–5% от всех опухолей желудочно-кишечного тракта. В США ежегодно регистрируется примерно 11 110 новых случаев рака тонкого кишечника и 1 700 смертельных исходов. Прочитайте дополнительные сведения и толстой кишки Колоректальный рак Колоректальный рак является чрезвычайно распространенным явлением. К клиническим проявлениям относятся наличие примеси крови в кале и изменения характера опорожнения кишечника. Для соответствующих. Прочитайте дополнительные сведения
На коже и слизистых выявляются гиперпигментированные пятна – в особенности, вокруг рта, на губах и деснах, кистях и стопах. Пятна могут разрешаться в пубертатном возрасте, за исключением располагающихся на слизистой щек. Полипы могут кровоточить и нередко служат причиной кишечной непроходимости или инвагинации.
Диагностика синдрома Пейтца-Егерса основана на клинической картине. Для этого синдрома следует оценить пациентов с периоральной или буккальной пигментацией и/или ≥ 2 GI гамартматозных полипов или семейную историю синдрома Пютца-Джегерса, включая тестирование мутаций STK11 (см. также Клинические рекомендации о генетическом тестировании и лечении наследственных синдромов рака желудочно-кишечного тракта Американский колледж гастроэнтерологии [American College of Gastroenterology's clinical guidelines about genetic testing and management of hereditary GI cancer syndromes]).
Профилактическое обследование на рак ЖКТ у пациентов с синдромом Пютца-Джегерса включает колоноскопию, эндоскопию верхних отделов пищеварительного тракта и капсульную эндоскопию, начиная с 8 лет с последующим наблюдением, определяемым результатами. Профилактическое обследование на рак молочной железы, яичников, эндометрия и шейки матки должно включать самообследование молочных желёз, начиная с 18 лет, а затем начиная с 25 лет включать ежегодное гинекологическое обследование, тазовое или трансвагинальное УЗИ, мазок по Папаниколау (ПАП-тест) и МРТ грудной клетки и/или маммографию. Кроме того, профилактическое обследование на рак поджелудочной железы должно начаться в возрасте 30 лет и включать магниторезонансную холангиопанкреатографию (МРХПГ) или эндоскопическую ультрасонографию. Профилактическое обследование семенников (на предмет опухоли клеток Сертоли) с помощью осмотра семенников должно проводиться ежегодно от рождения до подросткового возраста; УЗИ следует проводить в случае выявления аномалии при пальпации или при феминизации. Хотя пациенты с синдромом Пютца-Джегерса подвержены повышенному риску развития рака легких, не рекомендуется проводить специальный скрининг, но следует учитывать, если пациенты курят (см. также Клинические рекомендации о генетическом тестировании и лечении наследственных синдромов рака желудочно-кишечного тракта Американский колледж гастроэнтерологии [American College of Gastroenterology's clinical guidelines about genetic testing and management of hereditary GI cancer syndromes])
Родственников первой степени родства необходимо обследовать на предмет повреждений кожи, характерных для синдрома Пютца-Джегерса.
Полипы толстой кишки > 1 см, как правило, удаляют.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
учебники / 7-Синдром Пейтца
Основной признак этого наследственного синдрома — множественные мелкие пигментные пятна (лентиго) на губах и слизистой рта. Вторая составляющая синдрома — полипоз тонкой кишки, толстой кишки и желудка, который приводит к приступам боли в животе и желудочно-кишечным кровотечениям. Полипы представляют с обой гамартомы; они имеются у подавляющего большинства, но не у всех больных.
Синонимы: периорифициалъный лентигиноз, синдром Пейтца—Турена, полипоз пигментно - пятни стый.
Эпидемиология
Наследственность
Тип наследования — аутосомно-доминантный. Частота спонтанных мутаций составляет около 40%. Локус гена неизвестен.
Лентиго обнаруживают у грудных детей и детей младшего возраста; полипы — у детей старшего возраста и взрослых до 30 лет.
Лентиго бывает врожденным или появляется в грудном и младшем детском возрасте. Пятна на губах со временем могут исчезнуть, но пигментация слизистой рта сохраняется на всю жизнь — без этого признака нельзя поставить диагноз. Лентиго обнаруживают и в отсутствие полипоза ЖКТ.
Приступы боли в животе, желудочно-кишечные кровотечения, анемия. Приступы боли в животе начинаются в возрасте 10— 30 лет, реже — в более старшем возрасте. Больные предрасположены к раку молочной железы, яичников и поджелудочной железы.
Сходные симптомы выявляют и у других членов семьи.
Физикальное исследование
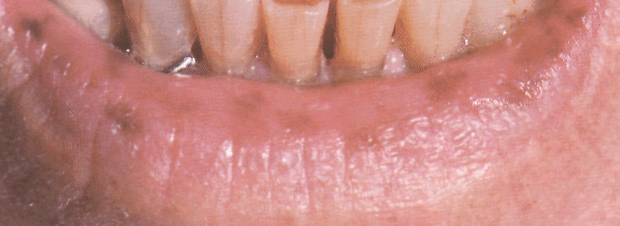
Элементы сыпи. Пятна. Цвет. Темно-коричневый или черный. Размеры. От 2 до 5 мм. Лентиго на лице — мельче, чем на ладонях, подошвах и слизистой рта.
Форма. Круглая или овальная. Расположение. Тесные скопления на губах, особенно на нижней , вокруг рта и на переносице.
Локализация. Губы, слизистая щек , окружность естественных отверстий, нос, подбородок, ладони и подошвы, тыльная поверхность кистей.
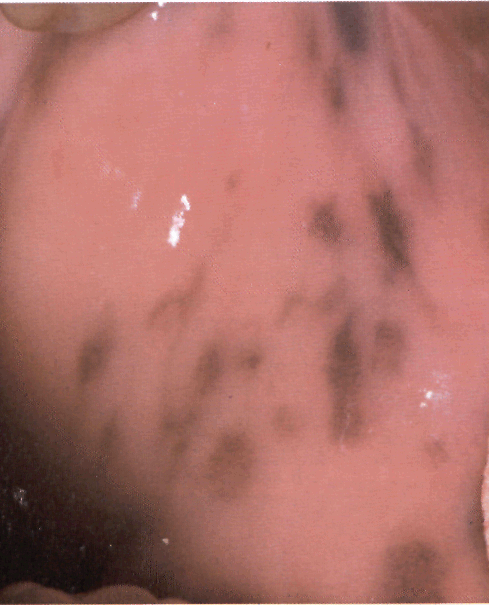
Обязательный признак синдрома Пейтца— Егерса — пятна коричневого, черного или иссиня-черного цвета, беспорядочно рассеянные на деснах, слизистой щек и твердого неба.
Гиперпигментированные полоски, изредка — диффузная гиперпигментация ногтевого ложа.
Полипы тонкой, реже — толстой кишки. Возможны инвагинация и обтурационная кишечная непроходимость. Из полипа может развиться аденокарцинома.
Дифференциальный диагноз
Гиперпигментированные пятна на коже Веснушки (светлее, чем лентиго при синдроме Пейтца—Егерса); старческое лентиго; множественные лентиго, первичная надпочечниковая недостаточность, Пигментация слизистых Конституциональная пигментация у представителей цветных рас, татуировка амальгамой, лечение зидовудином.
Рисунок Синдром Пеитца—Егерса. Множественные темно-коричневые лентиго на красной кайме нижней губы
Дополнительные исследования
Лентиго, Усиление синтеза меланина в эпидермисе (при локализации лентиго на ладонях и подошвах возможно нарушение передачи меланосом кератиноцитам). Усиленное накопление меланина в меланоцитах и клетках базального слоя эпидермиса. Полипы ЖКТ. Сочетание гладко-мышечных и железистых элементов.
Общий анализ крови
Возможна постгеморрагическая анемия.
Анализ кала на скрытую кровь позволяет выявить бессимптомные кровотечения.
У каждого больного с лентигинозом необходимо исследовать ЖКТ.
Анамнез, клиническая картина и выявление полипов ЖКТ.
Течение и прогноз
К преждевременной смерти может привести лишь вовремя не распознанная злокачественная опухоль ЖКТ. У японцев с синдромом Пейтца—Егерса злокачественные опухоли возникают особенно часто, поэтому им показана профилактическая колэктомия.
Если больной настаивает, лентиго можно удалить с помощью лазера.
Каждые 1—2 года больного должен осматривать гастроэнтеролог или хирург. Полипы, превышающие в размерах 1,5 см, а также кровоточащие, удаляют.
Рисунок . Синдром Пеитца—Егерса. На слизистой щек видны скопления темно-коричневых пятен
Лентигиноз периорифициальный
Заболевание рассматривается как нейро-мезенхимальная дисплазия, вызванная генной мутацией, передающаяся аутосомно-доминантно с плейотропным эффектом. Klostermann (1963), наблюдавший в семье 15 больных, относит этот синдром к факоматозам. Из 31 случая, проанализированных Touraine и Coder (1946), в 22 было несколько больных в семье в 3-4 поколениях.
Может быть значительная вариабельность и диссоциация признаков среди родственников. Например, у больной, наблюдавшейся Farmer и его соавторами (1963), был полный синдром, у её брата - только пигментации, у отца - полипоз. По поводу малосимптомных случаев Klostermann замечает, что при оценке этого явления должны учитываться даже минимальные изменения, недостаточные для постановки диагноза, но могущие быть связанными со специфическим эффектом гена, а также вариабельность временного интервала, в котором более характерные признаки появятся или, наоборот, исчезнут, как, например, пигментация. Кроме того, отсутствие клинически и рентгенологически полипоза ещё не исключает изменений в слизистой, которые могли проявляться в микроскопически малых папилломах. Подтверждением этому могут служить данные Р. В. Савиной и её соавторов (1983), обнаруживших гистологически гамартомные кисты в зоне макроскопически неизменённых участков слизистой оболочки желудка у больной с синдромом Пейтца-Егерса 18 лет, погибшей от рака желудка.
Спорадические случаи считаются проявлением новых мутаций.
Множественные мелкие пигментные пятна от светло-коричневого до чёрного цвета овальных или округлых очертаний, густо расположенные вокруг и в полости рта, на губах, особенно нижней, периназально, периорбитально, реже на конечностях (на ладонях и подошвах, тыльной поверхности пальцев). Описаны случаи с ограниченными высыпаниями в полости рта и на красной кайме губ. В полости рта могут быть и папилломатозные изменения (Lowe, 1975). А. В. Брайцев и Г. М. Большакова описали генерализованные лентигинозные высыпания. По данным Bartholomew, красная кайма губ поражается у 96% больных, слизистая щек - у 83%, кожа вокруг естественных отверстий лица - у 36%, конечностей - у 32%. Изменения могут быть и на роговице (Degos, 1981).
Заболевание ограничивается только кожными изменениями лишь у единичных больных, у большинства наблюдается сочетание с множественными полипами желудочно-кишечного тракта, особенно тонкого кишечника. Полипы могут быть и другой локализации: мочевой пузырь, почечные лоханки, мочеточник, бронхи, нос.
На основании анализа литературы Klostermann пришел к выводу, что в 33% случаев проявления полипоза развиваются в первом десятилетии жизни, 71% - к концу второго и 31% - третьего. Но эти сроки, по наблюдениям автора, могут быть значительно более поздними, с появлением кишечной симптоматики в 38 и даже 50-летнем возрасте.
Bandler (1950) описал семью, в которой у одного из членов семьи имелся множественный лентигиноз с локализацией на губах, периорально, на слизистой щек, а также на тыльной поверхности кистей и пальцев, в сочетании с множественными гемангиомами тонкого кишечника, но без полипов. У нескольких других родственников был множественный гемангиоматоз кишечника без лентигиноза. Автор ставит вопрос о взаимодействии генов, ответственных за развитие пигментации, кишечных полипов и гемангиом. У больных, описанных Торсуевым Н. А. (1973), наблюдалось сочетание с множественными пигментными, в том числе гигантскими, невусами.
Заболевание развивается в первые годы жизни, но может существовать с рождения. В редких случаях периорифициальный лентигиноз возникает у взрослых. С возрастом интенсивность пигментации на коже может изменяться, в полости рта она обычно остается без изменений.
Начиная с подросткового возраста или немного позже, начинает клинически проявляться полипоз желудка и кишечника: боли, рвота, кровотечение, симптомы непроходимости, инвагинации, вторичной анемии. Зачастую синдром выявляется из-за развившихся осложнений после хирургического вмешательства по поводу непроходимости или развития опухоли. Иногда больные подвергаются многократным операциям.
Наиболее неблагоприятным считается повышенный риск злокачественного перерождения полипов, преимущественно желудка, 12-перстной кишки, толстого кишечника. Данные о частоте развития карцином неоднородны. В частности, имеется мнение (Belisario), что полипы при этом синдроме представляют собой гамартомы и поэтому протекают доброкачественно. Castelman и Kruickstein нашли в 33 случаях из 57, диагностированных кач; карцинома на фоне полипов, аденоматозные полипы с очаговой атипией. Newbold, приводя данные Achord и Proctor о том, что висцеральный канцер развивается примерно у 20% всех больных лентигинозом периорифициальным, отмечает трудность истинной оценки частоты, так как неясно, все ли случаи, исследованные гистологически, являются злокачественными опухолями, а не гамартомами. Кроме того, в эту серию могли быть включены другие формы наследственных полипозов кишечника с большей потенциальностью к раковому перерождению. Более вероятными автор считает данные Asman и Pierse о 10%-ной или меньшей частоте развития рака в сравнении с 60% или более - при семейном полипозе.
По мнению Reid, при синдроме Пейтца-Егерса-Турена имеется повышенный риск развития рака кишечника, составляющий 2-3%. Опухоли чаще развиваются в зоне 12-перстной кишки. Неоплазма может возникнуть или на нормальной слизистой или из полипов, развившихся при этом синдроме. Что касается полипов толстого кишечника, то здесь может быть сочетание двух типов - гамартомных и аденоматозных, часто переходящих в карциному. Автор поддерживает точку зрения Olansky и Achord о том, что превентивное хирургическое вмешательство не показано и не выполнимо, но настороженность - обязательна.
Harnack полагает, что оперативное вмешательство в основном показано как средство профилактики инвагинаций. Nishiyama и его соавторы, описывая дифференциальную диагностику между синдромом Пейтса-Егерса и другими состояниями, протекающими с изменениями кожи и полипозом кишечника, отмечают в качестве одного из признаков, отличающих этот синдром, большую опасность возникновения инвагинаций, чем злокачественной опухоли. Указывается, что злокачественное перерождение с метастазами наблюдается чрезвычайно редко. К таким достоверным случаям автор относит только наблюдения Achord, Proctor и Horn.
По мнению Degos, злокачественное перерождение наблюдается в 2-5% случаев.
На основании анализа литературы Farmer и его соавторы отмечают, что наиболее частым является боль в животе, вызванная инвагинацией полипов (около 85%), затем кровотечение, анемия, общая слабость. Хирургическое вмешательство рекомендуется только при непроходимости или кровотечении, но не как мера профилактики, а радикальные или обширные хирургические вмешательства считаются нецелесообразными. С точки зрения автора, необходимо проводить тщательный дифференциальный диагноз между почти совершенно доброкачественным синдромом Пейтса-Егерса и предзлокачественным семейным полипозом толстого кишечника, что бывает трудно, если нет пигментаций или имеется выраженный полипоз толстого кишечника. Но даже, если при синдроме Пейтса-Егерса обнаруживается эта локализация полипов, это не является показанием для тотальной колонэктомии, как при семейном полипозе или синдроме Гарднера. Авторы считают, что полипы при этом синдроме являются гамартомами.
Newbold заключает: чем тяжелее полипозное поражение кишечника, тем больше частота изменений на коже и больше вероятность развития раковой опухоли. Burdick и Prior указывают на повышенный риск развития у больных также опухолей мочеполовых органов, лёгких, яичников, молочной железы.
Увеличение пигмента, количества меланоцитов в базальном слое, макрофагов, содержащих пигмент, в дерме. В некоторых зонах меланоциты могут выявляться в виде гнезд в зоне дермо-эпидермального соединения.
Дифференциальный диагноз
Дифференцировать лентигиноз периорифициальный необходимо от веснушек, различных форм генерализованного лентиго (Leopard-синдром и др.), центрофационального лентигиноза, гастроинтестинального полипоза (Cronkite-Canada синдром), болезни Аддисона. синдрома Gardner.
Вернуться к списку статей о кожных заболеваниях
Реклама, размещённая на сайте «Ваш дерматолог», является одним из источников его финансирования.
Наличие рекламы медицинских центров, лекарств, методов лечения, нельзя расценивать как рекомендацию владельца сайта к их посещению, приобретению или применению.
Последнее обновление страницы: 30.11.2014 Обратная связь Карта сайта
© NAU. При цитировании и копировании материалов убедительная просьба делать активную ссылку на сайт «Ваш дерматолог»
Представленная на сайте информация не должна использоваться для самостоятельной диагностики и лечения
и не может служить заменой очной консультации врача - дерматолога.
Читайте также: