Лучевые признаки синдрома Корнелии де Ланге (СКДЛ) у плода
Добавил пользователь Владимир З. Обновлено: 28.01.2026
а) Терминология:
1. Сокращения:
• Синдром Корнелии де Ланге (СКДЛ)
2. Синонимы:
• Синдром Брахманна-Ланге
3. Определения:
• Редкое мультисистемное заболевание, проявляющееся характерными чертами лица, задержкой роста, нарушением умственного развития, пороками развития конечностей, патологией ЖКТ, пороками сердца и гипертрихозом
б) Лучевая диагностика:
1. Общие сведения:
• Критерии диагностики:
о Выраженная ЗРП в сочетании с дефектами конечностей и аномалиями внутренних органов
(Слева) УЗИ в III триместре. Лицо плода с СКДЛ. Определяются тонкие губы, вытянутый губной желобок впалая нижняя челюсть, а также дефект конечности.
(Справа) Клиническая фотография новорожденного с СКДЛ. Отмечаются некоторые характерные признаки заболевания, включая длинные ресницы тонкие губы и вытянутый губной желобок. (Слева) УЗИ плода во II триместре, фронтальная плоскость. СКДЛ. Визуализируется правосторонняя диафрагмальная грыжа, печень находится в полости грудной клетки. Наличие диафрагмальной грыжи на фоне СКДЛ связано с крайне неблагоприятным прогнозом.
(Справа) Тот же случай. Фотография, полученная во время посмертного исследования. Определяется крупная правосторонняя ВДГ, печень занимает практически всю правую половину грудной клетки. Наличие ВДГ в сочетании с пороками развития конечностей позволяет заподозрить СКДЛ. (Слева) УЗИ плода в 14 нед. Патологические находки, позволяющие заподозрить диафрагмальную грыжу: дно желудка находится в грудной клетке, сердце смещено к левой стенке грудной клетки. Смещение желудка кзади позволяет предположить, что печень также находится в полости грудной клетки. Позднее у плода диагностирован СКДЛ.
(Справа) 3D УЗИ плода в III триместре. СКДЛ. Редукционный дефект конечности. Дефекты конечностей, как правило, двусторонние, нередко асимметричные, с поражением преимущественно верхних конечностей.
2. УЗИ при синдроме Корнелии де Ланге у плода:
• Редукционные дефекты верхних конечностей, часто тяжелые
• Микрогнатия, выступающая верхняя губа или губной желобок
• ВДГ: не является обязательным критерием для постановки диагноза
• Выраженная ЗРП, развивающаяся на ранних сроках
3. Рекомендации по лучевой диагностике:
• Советы по проведению исследования:
о При подозрении на СКДЛ выполняют 3D УЗИ с целью исследования анатомии лица и конечностей
в) Дифференциальная диагностика синдрома Корнелии де Ланге у плода:
1. Синдром Фринса:
• ВДГ (89%), гипоплазия дистальных сегментов конечностей (75%), грубые черты лица
• Многоводие, рост плода в норме
2. Анеуплоидия:
• Синдром Паллистера-Киллиана:
о Соматический мозаицизм с наличием изохромосомы 12р (мозаичная тетрасомия 12р)
о ВДГ, многоводие
о Ризомелическое укорочение конечностей, редко - гипоплазия дистальных сегментов конечностей
• Частичная дупликация 3q:
о Краниосиностоз, пороки развития сердца и почек о Рост плода в норме; отставание темпов роста в постнатальном периоде
о Низко расположенная передняя линия роста волос, густые брови, длинные ресницы
• Т18:
о ЗРП, дефекты радиального луча, скрещенные пальцы
о ВДГ (редко)
3. Фетальный алкогольный синдром:
• Задержка роста в пре- и постнатальном периоде
• Микроцефалия, пороки сердца, отставание в развитии
• Короткая глазная щель, сглаженный губной желобок, тонкая верхняя губа
4. Изолированная ВДГ:
• Проводят тщательный поиск других пороков развития, в частности сердца
5. Редукционные дефекты конечности:
• Изолированные или в составе синдрома
г) Патологоанатомические особенности. Общие сведения:
• Этиология:
о Мутация одного из трех когезиновых белков (60-65% случаев):
- NIPBL: регулятор функции когезинов и человеческий гомолог nipped-B, присутствующего у дрозофилы
- SMC1A и SMC3: кодируют компоненты когезинового кольца
- Когезин регулирует когезию сестринских хроматид в ходе митоза и мейоза; играет важную роль в регуляции экспрессии генов
• Генетические факторы:
о Аутосомно-доминантный тип наследования
о В большинстве случаев (99%) мутация возникает случайно; возможно семейное наследование (редко)
(Слева) Характерные черты лица мертворожденного с СКДЛ. Гирсутизм В, тонкие брови. Ресницы характерно удлинены. Антимонголоидный разрез глаз и горизонтальные складки под нижними веками. Губы тонкие, губной желобок удлинен, определяется микрогнатия.
(Справа) Схожие фенотипические проявления у другого ребенка с СКДЛ. Особенно заметны тонкие «подведенные» брови и длинные ресницы. Отмечаются асимметричные тяжелые дефекты конечностей. (Слева) Плод с СКДЛ в III триместре. 3D УЗИ позволяет визуализировать фенотипические проявления синдромов, характеризующихся множественными аномалиями развития. Отчетливо визуализируются выступающее надпереносье, удлиненный губной желобок и микрогнатия. В конце III триместра могут также определяться удлиненные ресницы.
(Справа) Тот же новорожденный в профиль. Ушные раковины выглядят увеличенными за счет небольших размеров головки. Отмечаются удлиненный желобок и микрогнатия. (Слева) Фотография доношенного мертворожденного с СКДЛ и выраженной микрогнатией И Определяются довольно типичная монодакти-лия, один из вариантов редукционных дефектов конечности, а также мясистый вырост кожи.
(Справа) УЗИ верхней конечности плода во II триместре. СКДЛ. Обратите внимание на характерный дефект, монодактилию Пальцы и трубчатые кости дистального сегмента противоположной конечности отсутствуют. Подобная асимметрия дефектов конечностей крайне характерна для СКДЛ.
д) Клинические особенности:
1. Клиническая картина:
• Характерные черты лица:
о Густые дугообразные брови («подведенные»); вытянутый сглаженный губной желобок; тонкие губы; серповидный рот; синофриз (сросшиеся брови); длинные ресницы; птоз; антимонголоидный разрез глаз; запавшая переносица; ноздри, вывернутые кпереди; микрогнатия
• Важный, но вариабельный признак - пороки развития конечностей:
о От укорочения плечевых костей или уменьшения размеров кистей до тяжелых редукционных дефектов конечностей и монодактилии
• Микробрахицефалия, короткая шея, низко расположенная задняя линия роста волос, передняя линия роста волос проходит на лбу
• ЗРП, карликовость в постнатальном периоде
• Пороки развития сердца (25%): самые частые - стеноз легочной артерии, ДМЖП
• Диафрагмальная грыжа: прогностический признак неблагоприятного исхода
• Другие аномалии пищеварительного тракта: незавершенный поворот кишечника, удвоение толстой кишки, заворот слепой кишки, пилоростеноз, рефлюксная болезнь
2. Демографические особенности:
• Заболеваемость достигает 1:10 000 родов
3. Естественное течение и прогноз:
• Степень тяжести широко варьирует:
о От смертельных случаев в перинатальном периоде до умеренных проявлений, позволяющих пациенту обслуживать себя самостоятельно во взрослом возрасте
• Нарушения умственного развития (от умеренных до выраженных)
• Выраженная задержка речевого и вербального развития, тугоухость, судороги (11-23%)
• Поведенческий фенотип: склонность к членовредительству, агрессивность, нарушение сна, аутистическое поведение
е) Особенности диагностики. Признаки, учитываемые при интерпретации результатов:
• Наличие ВДГ в сочетании с пороками развития конечностей позволяет заподозрить СКДЛ
ж) Список использованной литературы:
1. Kline AD et al: Clinical, developmental and molecular update on Cornelia de Lange syndrome and the cohesin complex: Abstracts from the 2014 Scientific and Educational Symposium. Am J Med Genet A. 167(6): 1179-92, 2015
2. Muto A et al: Nipbl and mediator cooperatively regulate gene expression to control limb development. PLoS Genet. 10(9):el004671, 2014
Редактор: Искандер Милевски. Дата обновления публикации: 17.11.2021
Синдром Эдвардса
Общие сведения
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причины синдрома Эдвардса
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета - скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения - олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Синдром Корнелии де Ланге ( Амстердамская карликовость , Амстердамский нанизм , Синдром Брахмана де Ланге )
Синдром Корнелии де Ланге – это генетически опосредованное мультисистемное заболевание, включающее множественные аномалии развития и олигофрению. Фенотипические признаки синдрома представлены микробрахицефалией, заниженной линией роста волос, тонкими сросшимися бровями, широкой запавшей переносицей, микрогенией и др. Характерна низкорослость, возможны врожденные пороки (ВПС, мочеполовые аномалии, пилоростеноз, диафрагмальная грыжа). При диагностике учитываются клинические критерии и результаты генетического тестирования. Дети с данной патологией нуждаются в симптоматической терапии, дефектологической помощи.
МКБ-10
Синдром, характеризующийся комплексом стигм дизэмбриогенеза, пороками развития и умственной неполноценностью, был описан в 1916 г. доктором медицины В. Брахманом. Однако свое официальное название заболевание получило в честь педиатра из Амстердама Корнелии де Ланге, которая подробно описала сразу два клинических наблюдения в 1933 г. В литературе наряду с общепринятым – синдром Корнелии де Ланге – встречаются названия «синдром Брахмана де Ланге», «амстердамская карликовость/нанизм». Патология регистрируется с частотой 1:10000–1:30000, распространенность не имеет географических, расовых и гендерных различий.
Причины
Синдрому свойственна генетическая неоднородность, на данный момент известно три гена, ответственных за данную патологию. Около 50% описанных случаев связано с дефектами в гене NIPBL, кодирующем белок делангин (5p13.2), около 5% – с мутациями гена когезинового комплекса SMC1A (Xp11.22). Один известный случай ассоциирован с нарушением другой субъединицы когезина, кодируемой SMC3 (10q25). Примерно в 40% наблюдений синдром Корнелии де Ланге вызывается неизвестными мутациями, которые еще только предстоит вычислить.
Большая часть мутаций NIPBL являются вновь возникшими, поэтому чаще больные дети рождаются от генетически здоровых родителей. Однако известны наблюдения семейных случаев с аутосомно-доминантным наследованием. Ген SMC1A расположен на половой Х-хромосоме – при его дефектах наследование сцеплено с полом (болеют мужчины, женщины выступают гетерозиготными носительницами).
Факторы риска
Спонтанные патологические изменения генетического материала могут быть обусловлены следующими факторами:
- интоксикациями и инфекциями беременной, перенесенными в первом триместре;
- эндокринопатиями беременной;
- лекарственным (в т. ч. цитостатическим), радиационным воздействием на плод;
- близкородственным браком;
- поздними родами (у женщин старше 35 лет).
При отсутствии генных мутаций у родителей риск повторного рождения ребенка с синдромом Брахмана де Ланге в одной семье равен 2-5%.
Патогенез
Молекулярную основу патологии составляет нарушение функционирования белков когезинового комплекса. Когезины играют важнейшую роль в процессе клеточного деления. Они располагаются внутри хромосомы, сцепляя и удерживая между собой две сестринские хроматиды. Другие, не менее важные функции когезина – это репарация ДНК и регуляция экспрессии генов.
По всей видимости, воздействие различных мутагенных факторов в критические периоды приводит к неправильному делению клеток и закладке органов. Патологоанатомическое исследование выявляет значительные изменения головного мозга: аплазию оперкулярной коры, недоразвитие роландовой борозды, запаздывающую миелинизацию и и миелодегенерацию. Также обнаруживаются пороки развития почек, надпочечников, гонад, сердца, гипоплазия тимуса.
Классификация
По степени выраженности фенотипических проявлений выделяют 2 варианта синдрома Корнелии де Ланге. Они различаются как проявленностью внешней симптоматики, так и витальным прогнозом:
- классический: значительная задержка развития плода, грубые пороки развития и выраженная умственная неполноценность;
- доброкачественный: лицевые и скелетные аномалии без грубых пороков внутренних органов, пограничная ЗПР. Дефекты генов SMC3 и SMC1A коррелируют с доброкачественным течением синдрома.
Симптомы
Физическое развитие
Младенцы с синдромом Корнелии де Ланге рождаются с низкими росто-весовыми показателями: средняя масса составляет 2100-2300 г. Они малоактвны, редко выражают свои потребности криком. С первых дней отмечается вялое сосание груди, частые срыгивания, диспепсические расстройства. Такие проблемы нередко вынуждают кормить ребенка через назогастральный зонд или гастростому. Дети часто страдают рецидивирующими респираторными инфекциями. Физическое и психомоторное развитие отстает от возрастной нормы.
Фенотипические признаки
Внешний вид ребенка очень своеобразный. Перечисленные черепно-лицевые дисморфии входят в число основных критериев при диагностике патологии:
- аномалии черепа и лицевого скелета: микро- и брахицефалия, готическое небо, небные расщелины, микрогнатия;
- черты лица: тонкие сросшиеся брови, длинные ресницы, широкая переносица с седловидной деформацией и широкими ноздрями, низко посаженные уши, тонкие губы с опущенными вниз уголками;
- аномалии кожи и ее придатков: на голове ‒ густые волосы с низкой линией роста, на теле – гипертрихоз, мраморная кожа.
Скелетные аномалии
Пациенты имеют воронкообразную грудную клетку, короткую шею. В области позвоночного столба могут обнаруживаться такие дефекты, как Spina bifida, люмбализация или сакрализация позвонков. Присутствует гипоплазия кистей и стоп, дефекты развития пальцев (клинодактилия, синдактилия, олигодактилия), суставные контрактуры, дисплазия и врожденный вывих бедра. Отставание в росте сохраняется и во взрослом возрасте: мужчины с амстердамским нанизмом имеют рост около 156 см, женщины ‒ 131 см.
Неврологические и психопатологические проблемы
Больные с классической формой синдрома Корнелии де Ланге имеют интеллектуальные нарушения в степени имбецильности или глубокой дебильности. При мягкой форме интеллектуальный дефект выражен нерезко – больные имеют диагноз ЗПР. У четверти пациентов эпизодически отмечается судорожный синдром.
Поведенческие особенности представлены синдромом гиперактивности, тревожностью, признаками ОКР. Отмечается агрессивность по отношению к окружающим, аутоагрессия и самоповреждающее поведение, стереотипные действия. Речевые расстройства носят системный характер, страдает вербальная коммуникация, при небных расщелинах возникает ринолалия.
Зрительные нарушения
Распространенными проблемами со стороны зрительной системы выступают миопия, птоз века, блефарит. Реже диагностируется косоглазие, микрокорнеа, в единичных наблюдениях выявлены катаракта и глаукома. При тяжелом варианте синдрома Корнелии де Ланге высок риск отслойки сетчатки и атрофии зрительного нерва.
Пороки внутренних органов
Почти в половине случаев синдрому сопутствуют врожденные кардиальные пороки: дефекты перегородок сердца (аортолегочной, межжелудочковой, межпредсердной), коарктация аорты, тетрада Фалло. Аномалии развития ЖКТ чаще всего представлены гастроэзофагеальным рефлюксом (90%), может выявляться диафрагмальная грыжа (2%), пилоростеноз (4%), незавершенный поворот кишечника (10%). Частыми патологиями мочеполовой системы выступают поликистозная или подковообразная почка, гидронефроз, двурогая матка, неопущение яичек, гипоспадия. Характерна задержка полового развития.
Осложнения
Тяжелые варианты синдрома ассоциированы с пороками развития, несовместимыми с жизнью, и гибелью новорожденных вскоре после рождения. При более мягком течении тормозить развитие ребенка могут рецидивирующие синуситы, отиты с последующим развитием кондуктивной тугоухости. На фоне гастроэзофагеального рефлюкса нередко возникает анемия, аспирационная пневмония, стеноз пищевода, иногда − пищевод Барретта, аденокарцинома. В бытовом плане пациенты с синдромом Корнелии де Ланге нуждаются в уходе, всесторонней помощи. Им требуется специальное обучение и дефектологическое сопровождение.
Диагностика
При рождении диагностическая гипотеза о наличии у ребенка синдрома Корнелии де Ланге может быть выдвинута на основании визуально определяемых дефектов. Новорожденный должен быть осмотрен неонатологом, детским кардиологом, хирургом, неврологом. В плановом порядке проводятся консультации челюстно-лицевого хирурга, эндокринолога, ортопеда, логопеда. Диагностическая тактика включает:
- Антропометрию. При рождении и на протяжении всего раннего детства проводится регулярное измерение длины (роста), массы тела, окружности головы. Все антропометрические показатели находятся ниже возрастных норм (
- Обследование сердечно-сосудистой системы. Выявить наличие сердечных пороков позволяет ЭхоКС, ЭКГ. При необходимости могут потребоваться инвазивные исследования: катетеризация сердца, вентрикулография и др.
- Обследование ЖКТ. Для обнаружения ГЭРБ и морфологических изменений пищевода показана ЭГДС под наркозом. С целью исключения возможных аномалий кишечника необходима рентгенография с барием.
- Психоневрологическую диагностику. При первичном осмотре определяются рефлексы, тонус мышц, уровень психомоторного развития. В дальнейшем исследуется НСГ, ЭЭГ, выполняется рентгенография костей черепа, МРТ головного мозга.
- Прочие обследования. В целях выявления аномалий органов осуществляется УЗИ брюшной полости, почек, гениталий. Обязательна проверка функции зрения (рефрактометрия, офтальмоскопия, биомикроскопия), слуха (отоскопия, аудиограмма, слуховые ВП). С помощью рентгена костей и позвоночника выявляются скелетные аномалии.
- Молекулярное тестирование. Генодиагностика синдрома предполагает консультирование семьи генетиком с проведением кариотипирования больного ребенка, поиском мутаций в генах NIPBL, SMC1A.
Выявление синдрома возможно на дородовом этапе в рамках пренатального УЗИ-скрининга и фетометрии плода. Эхографическими маркерами патологии служат ЗВУР, особенности черепа, аномалии конечностей. С помощью УЗИ могут быть обнаружены ВПС, диафрагмальная грыжа и другие аномалии органов плода.
Дифференциальная диагностика
При проведении обследования необходимо исключить другие заболевания, имеющие сходные фенотипические черты:
- алкогольный синдром плода;
- трисомию 3q;
- синдром Фринса;
- синдром Нунан;
- синдром Прадера-Вилли;
- синдром Рубинштейна-Тейби.
Лечение синдрома Корнелии де Ланге
Тактика зависит от комплекса выявленных пороков, степени их влияния на качество жизни ребенка, его социализацию. В первую очередь осуществляется лечение тех патологий, которые могут привести к дестабилизации состояния:
- Желудочно-пищевой рефлюкс: разработка диеты, прием антацидов и H2-гистаминоблокаторов, по показаниям – фундопликация.
- Пороки сердца: ушивание/пластика дефектов перегородок, дилатация или резекция коарктации аорты, коррекция тетрады Фалло.
- Неврологические нарушения: прием противосудорожных препаратов, анксиолитиков, СИОЗС, лечение средой.
- Аномалии мочеполовой системы: орхипексия, коррекция гипоспадии, пластика гидронефроза, коррекция пузырно-мочеточникового рефлюкса.
- Расщелины неба: уранопластика, велофарингопластика.
- Скелетные аномалии: лечебная физкультура, массаж, физиотерапия с целью предотвращения суставных контрактур.
Коррекция тугоухости проводится с помощью слуховых аппаратов, аномалий зрения – с использованием очков, аппаратных методик. В программу педагогического сопровождения ребенка обязательно включаются занятия с дефектологом, психологом, логопедом. Терапия гормоном роста неэффективна.
Прогноз и профилактика
Ввиду большого количества аномалий развития средняя продолжительность жизни лиц с синдромом Корнелии де Ланге ниже, чем в популяции. Однако при благоприятном варианте пациенты могут дожить до 50-60 лет. Больным необходима социально-психологическая поддержка, трудовая реабилитация, помощь в самообслуживании. Вследствие имеющегося иммунодефцита критически важным является предупреждение инфекций.
Профилактика генетического синдрома требует исключения кровнородственных браков и ответственного планирования беременности. Беременным обязательно прохождение лабораторного и ультразвукового скрининга, а при необходимости – инвазивной пренатальной диагностики.
2. Синдром Корнелии де Ланге: клиника, диагностика, лечение (случай из практики)/ Бугаенко О.А., Сиротченко Т.А., Бондаренко Г.Г., Вельковская М.М.// Медицинский вестник Юга России. – 2018.
Синдром Клайнфельтера
Синдром Клайнфельтера - хромосомная патология, обусловленная наличием в мужском кариотипе одной или нескольких дополнительных женских половых хромосом. Синдром Клайнфельтера характеризуется первичным гипогонадизмом, маленькими размерами тестикул, бесплодием, гинекомастией, неглубоким снижением интеллекта. Решающая роль в диагностике синдрома Клайнфельтера принадлежит кариотипированию; также проводится анализ фенотипических признаков, определение полового хроматина, экскреции фолликулостимулирующего гормона с мочой, спермограмма и пр. Лечение при синдроме Клайнфельтера включает гормональную терапию, возможно - оперативную коррекцию гинекомастии, однако полное излечение синдрома невозможно.
Синдром Клайнфельтера – дисомия или полисомия по женской половой хромосоме, при которой у лиц мужского пола имеется не менее двух Х-хромосом и одна Y-хромосома. Синдром Клайнфельтера встречается с частотой 1 случай на 850-1000 новорожденных мальчиков. Среди детей, страдающих олигофренией, распространенность синдрома Клайнфельтера составляет 1–2%. Синдром получил название по фамилии американского врача Гарри Клайнфельтера, впервые описавшего его в 1942 г. Кариотип таких больных с дополнительной Х-хромосомой был определен в 1959 г. Поскольку ведущим клиническим проявлением синдрома Клайнфельтера является первичный гипогонадизм, ведением таких пациентов занимаются специалисты в области эндокринологии и андрологии.
Причины синдрома Клайнфельтера
Как и в случае синдрома Дауна, хромосомная аберрация при синдроме Клайнфельтера связана с нерасхождением хромосом (в последнем случае – половых) в процессе мейоза либо нарушением деления зиготы. При этом значительно чаще (в 60%) мальчики с синдром Клайнфельтера получают лишнюю материнскую Х-хромосому, чем отцовскую.
Среди возможных причин подобного рода хромосомных аномалий называются вирусные инфекции, поздняя беременность, неполноценность регуляторных механизмов материнской и отцовской иммунной системы.
При наличии лишней X-хромосомы развивается аплазия эпителия яичек, их последующая гиалинизация и атрофия, что во взрослом возрасте сопровождается азооспермией и эндокринным бесплодием. Среди причин мужского бесплодия синдром Клайнфельтера составляет 10%, о чем всегда должны помнить специалисты в области репродуктивной медицины.
Наиболее частым цитогенетическим типом является полный вариант синдрома Клайнфельтера с кариотипом 47,ХХY. Реже встречается мозаицизм (46XY/47XXY; 46XX/47XXY), еще реже – полисомия 48,XXXY; 48,XXYY; 49,XXXXY и т. д. При мозаичном варианте (около 10% случаев) часть клеток имеет нормальный кариотип, поэтому мужчины с синдром Клайнфельтера могут иметь нормально развитые и функционирующие половые железы и сохранные репродуктивные способности.
Симптомы синдрома Клайнфельтера
Ребенок с синдромом Клайнфельтера рождается с нормальными росто-весовыми показателями, правильной дифференцировкой наружных гениталий, обычными размерами тестикул. В раннем возрасте у мальчиков с синдромом Клайнфельтера может отмечаться частая заболеваемость ОРВИ, бронхитом, пневмониями. Такие дети обычно отстают в моторном развитии (позднее начинают держать головку, сидеть, стоять, ходить), имеют задержку речевого развития. Уже в возрасте 5-8 лет мальчики с синдромом Клайнфельтера отличаются высоким ростом, диспропорциональным телосложением (длинными конечностями, высокой талией). В допубертатном возрасте может обнаруживаться одно или двусторонний крипторхизм.
Умственная отсталость умеренной степени, трудности установления контакта со сверстниками, нарушения поведения отмечаются у половины больных синдромом Клайнфельтера.
Отчетливые внешние признаки, свидетельствующие о наличии у ребенка синдрома Клайнфельтера, проявляются в препубертатном и пубертатном периодах развития. К ним относятся евнухоидный тип телосложения, позднее появление вторичных половых признаков, гипоплазия яичек, малый половой член, гинекомастия. В постпубертатном периоде онтогенеза наблюдается инволюция тестикул, сопровождающаяся потерей фертильности. При осмотре подростка с синдромом Клайнфельтера выявляется отсутствие или скудный рост волос на лице и в подмышечных впадинах, оволосение на лобке по женскому типу. У большинства больных присутствуют редкие поллюции, эрекция, сохранно половое влечение, однако из-за выраженного андрогенного дефицита в среднем к 30 годам происходит снижение либидо и развивается импотенция.
Синдрому Клайнфельтера часто сопутствуют аномалии скелета (деформации грудной клетки, остеопороз), нарушения прикуса, врожденные пороки сердца и др. Характерно преобладание ваготонических реакций: брадикардии, акроцианоза, потливости ладоней и стоп. Со стороны органа зрения нередко отмечается нистагм, астигматизм, птоз века.
Больные с синдромом Клайнфельтера предрасположены к развитию сопутствующих заболеваний: эпилепсии, рака молочной железы, сахарного диабета, ХОБЛ, желчнокаменной болезни, варикозного расширения вен, ожирения, гипертонической болезни, ИБС, ревматоидного артрита, острого миелоидного лейкоза. Могут отмечаться психические заболевания - маниакально-депрессивный психоз, шизофрения и др. Есть данные, подтверждающие склонность больных с синдромом Клайнфельтера к алкоголизму, наркомании и гомосексуализму.
Диагностика синдрома Клайнфельтера
Как и другие хромосомные аномалии, синдром Клайнфельтера у плода может быть обнаружен еще на этапе беременности при проведении инвазивной пренатальной диагностики (амниоцетеза, биопсии хориона или кордоцентеза с последующим анализом кариотипа или КФ-ПЦР).
Постнатальная диагностика синдрома Клайнфельтера проводится эндокринологами, андрологами и генетиками. При исследовании полового хроматина в клетках слизистой оболочки полости рта присутствуют тельца Бара, что является маркером синдрома Клайнфельтера. Другими характерными признаками служат особые изменения кожного рисунка на пальцах. Тем не менее, окончательный диагноз хромосомной аномалии может быть установлен только после исследования кариотипа.
УЗИ мошонки выявляет уменьшение объема яичек. При исследовании андрогенного профиля уровень тестостерона в крови больных синдромом Клайнфельтера понижен, однако при этом отмечается повышение уровня фолликулостимулирующего и лютеинизирующего гормонов. При анализе спермограммы выявляется олиго- или азооспермия. Морфологическое исследование материала, полученного путем биопсии яичек, выявляет гиалиноз семенных канальцев, гиперплазию клеток Лейдига, уменьшение числа клеток Сертоли, отсутствие сперматогенеза.
В течение жизни мужчины с синдромом Клайнфельтера могут обращаться к андрологу, сексологу, эндокринологу с проблемами бесплодия, импотенции, гинекомастии, остеопороза и др., однако нередко основное заболевание так и остается нераспознанным.
Лечение синдрома Клайнфельтера
Полностью излечиться от синдрома Клайнфельтера не представляется возможным. Тем не мене, все больные нуждаются в проведении симптоматической и патогенетической терапии. В детском возрасте необходима профилактика инфекционных заболеваний, закаливание, занятия ЛФК, коррекция нарушений речи с помощью логопеда.
С подросткового возраста пациентам с синдромом Клайнфельтера назначается пожизненная заместительная терапия половыми гормонами (внутримышечные инъекции тестостерон-пропионата, сустанона-250; сублингвальный прием метилтестостерона и др.). Ранняя и адекватная гормонотерапия препятствует атрофии яичек, способствует повышению полового влечения, развитию вторичных половых признаков. При резко выраженном увеличении молочных желез проводится операция по коррекции гинекомастии.
С целью повышения трудоспособности и социальной адаптации, предупреждения психопатизации личности и ее асоциальной направленности показана психотерапия.
Прогноз и профилактика синдрома Клайнфельтера
Пациенты с синдромом Клайнфельтера имеют нормальную продолжительность жизни, однако склонность к развитию хронических заболеваний может стать риск-фактором ранней смертности. Большинство больных с синдромом Клайнфельтера бесплодны; единственно возможным вариантом рождения детей в семьях, где партнер болен, является использование донорской спермы. Тем не менее, при мозаичной форме синдрома Клайнфельтера мужчины могут стать отцами самостоятельно или воспользовавшись вспомогательными репродуктивными технологиями (ЭКО).
Для оценки вероятности рождения ребенка с синдромом Клайнфельтера в процессе ведения беременности женщинам предлагается прохождение пренатального скрининга. Однако даже в случае получения положительных данных за наличие синдрома Клайнфельтера у плода настаивание на прерывании беременности со стороны акушера-гинеколога является недопустимым. Решение вопроса о целесообразности пролонгирования беременности должно приниматься родителями. При нормальном кариотипе родителей риск повторного появления ребенка с такой же хромосомной аномалией составляет не более 1%.
Диспансерное наблюдение больных с синдромом Клайнфельтера осуществляется эндокринологом.
Лучевые признаки синдрома Корнелии де Ланге (СКДЛ) у плода
1. Сокращения:
• Муковисцидоз (МВ)
2. Определения:
• Мультисистемное заболевание с аутосомно-рецессивным типом наследования, вызванное нарушением транспорта ионов хлора через эпителиальные ткани
1. Общие сведения:
• Критерии диагностики:
о Во II триместре определяется гиперэхогенный кишечник, который к III триместру часто прогрессирует до дилатации кишечника
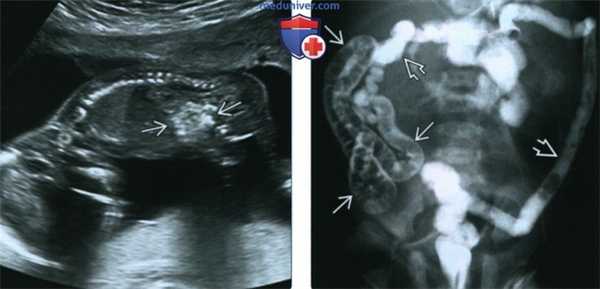
(Слева) УЗИ плода в 17 нед., сагиттальная плоскость. Определяются гиперэхогенные петли кишечника («яркие, как кость»). В случае выявления необходимо исключить все причины гиперэхогенности кишечника, включая муковисцидоз (МВ), анеуплоидию, инфицирование, пороки развития кишечника и кровотечение в анамнезе (заглатывание крови).
(Справа) Препятствием для прохождения контрастного вещества в данном случае является меконий. Определяются микроколон, а также многочисленные дефекты наполнения из-за скопления мекония в дистальном участке подвздошной кишки. Данная картина характерна для мекониевого илеуса при муковисцидозе (МВ).
2. УЗИ при муковисцидозе у плода:
• Гиперэхогенный кишечник во II триместре:
о Эхогенность кишечника превышает эхогенность костной ткани (патология)
о Усиление эхогенности связывают со скоплением слизи в просвете кишечника
о Величина риска МВ у плода с гиперэхогенным кишечником, по данным различных исследований, варьирует:
- По результатам крупного исследования, МВ диагностировали в 9,9% случаев гиперэхогенного кишечника
• Мекониевый илеус:
о Расширенный, гиперэхогенный тонкий кишечник
о Ультразвуковая картина может быть неотличима от таковой при атрезии подвздошной кишки
• Перфорация кишечника с мекониевым перитонитом:
о У 8% плодов с мекониевым перитонитом присутствует МВ
в) Дифференциальная диагностика муковисцидоза у плода:
1. Другие причины гиперэхогенности кишечника:
• Анеуплоидия, в частности Т21:
о Находят сопутствующую патологию, а также «малые» маркеры
• Инфицирование:
о Чаще всего - ЦМВ
• Ишемия кишечника
• Заглатывание крови
2. Атрезия подвздошной кишки:
• Ультразвуковая картина может быть неотличима от таковой при МВ
г) Патологоанатомические особенности. Общие сведения:
• Этиология:
о Мутации гена, кодирующего трансмембранный регулятор MB (CFTR):
- В настоящее время известно >1700 мутаций
- Генетические факторы
о Аутосомно-рецессивный тип наследования (риск повторного возникновения - 25%)
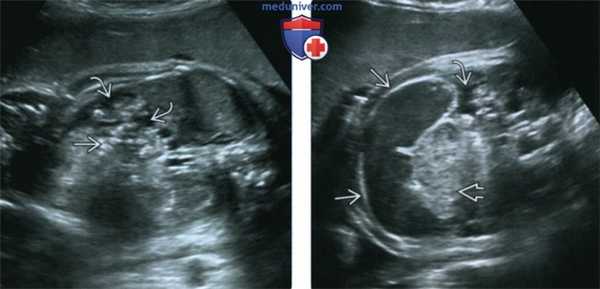
(Слева) УЗИ плода в 33 нед. Расчетный риск муковисцидоза (МВ) - 25%. Определяются незначительный асцит и умеренно гиперэхогенные петли кишечника.
(Справа) Тот же случай. Визуализируются признаки перфорации кишечника с мекониевым перитонитом: петля расширенного кишечника асцит, гиперэхогенный кишечник. В постнатальном периоде подтвержден диагноз муковисцидоза (МВ) и мекониевого перитонита. Несмотря на возможное совпадение ультразвуковой картины с атрезией подвздошной кишки, расширение дистального отдела тонкого кишечника следует всегда рассматривать как диагностический признак муковисцидоза (МВ).
1. Клиническая картина:
• Исследование на МВ входит в программу неонатального скрининга:
о Способствует ранней диагностике и своевременному лечению, позволяющему предотвратить развитие тяжелого дефицита питательных веществ
• У новорожденных заболевание может впервые проявиться задержкой отхождения мекония или отсутствием прибавки массы тела
• Преимущественное поражение дыхательной системы:
о Рецидивирующие инфекции, образование слизистых пробок
о Бронхоэктазия, гиперинфляция легких, поликистоз, спонтанный пневмоторакс
о Полипы носа, синуситы
• Поражение ЖКТ:
о Мальабсорбция вследствие недостаточности поджелудочной железы; СД (пожизненный риск - 40-50%)
• Мужское бесплодие вследствие врожденного отсутствия семявыносящих протоков
о Семявыносящие протоки закупорены слизью
2. Демографические особенности:
• 1:2500 родов среди неиспаноязычных белых; МВ страдают 30 000 детей и взрослых в США (в мире - 70 000)
• Уровень носительства: евреи-ашкеназы - 1/24; неиспаноязычные белые - 1/25; американцы азиатского происхождения -1/94
• Заболеваемость достигает максимума среди североевропеоидов:
о Самая частая мутация гена CFTR, delta F508, присутствует у 85% европеоидов с МВ
3. Естественное течение и прогноз:
• Медианная продолжительность жизни в настоящее время составляет 37,4 года, хотя прогнозируемая продолжительность жизни продолжает увеличиваться:
о Ожидаемая продолжительность жизни для нынешних детей составит более 40 лет; при сохранной функции поджелудочной железы она может достигать более 50 лет
4. Лечение муковисцидоза у плода:
• Гиперэхогенный кишечник по УЗИ → скрининг родителей на генетические мутации
• Для обнаружения мутации у плода выполняют амниоцентез:
о Неинвазивные методы диагностики находятся на стадии разработки
• Генетическое консультирование:
о При наличии в анамнезе установленной мутации во время последующих беременностей могут быть предложены биопсия ворсин хориона или преимплантационная генетическая диагностика
о Стандарт медицинской помощи: скрининг на муковисцидоз (МВ) предлагается всем беременным женщинам или женщинам, планирующим беременность:
- Чувствительность скрининга в различных этнических группах варьирует: от
е) Особенности диагностики. Важно знать:
• Обычное УЗИ не позволяет исключить муковисцидоз (МВ):
о Гиперэхогенный кишечник или мекониевый илеус определяются только в 11% случаев
• Отрицательные результаты скрининга на наличие мутаций не позволяют исключить носительство:
о Отрицательные результаты скрининга для неиспаноязычных белых (априорный риск - 1/25) означают наличие остаточного риска носительства величиной 1/200
• Ультразвуковая картина мекониевого илеуса может быть неотличима от таковой при атрезии подвздошной кишки: о Во всех случаях кишечной непроходимости у плода предлагают клиническое обследование на МВ
Видео этиология, патогенез муковисцидоза
Читайте также:
- Клиника и проявления кампилобактериоза. Лечение и профилактика кампилобактериоза
- КТ, МРТ саркомы Юинга позвоночника
- Оптимальная ориентация сегментарной пломбы при отслойке сетчатки. Рекомендации
- Лучевая диагностика сиалолита околоушной железы
- Патогенез алкогольной гипертензии. Механизмы развития гипертонии при алкоголизме