Микроцефалия на МРТ
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Микроцефалия – недоразвитие черепа и головного мозга, сопровождающее умственной отсталостью и неврологическими отклонениями. Микроцефалия характеризуется малыми размерами черепа, ранним смыканием черепных швов и закрытием родничка, судорожным синдромом, задержкой моторного развития, интеллектуальным дефектом, недоразвитием или отсутствием речи. Диагностика микроцефалии основывается на данных антропометрии, краниографии, КТ и МРТ головного мозга, ЭЭГ, НСГ; возможно пренатальное выявления микроцефалии у плода. При микроцефалии проводится симптоматическое лечение и реабилитационные мероприятия, направленные на социализацию ребенка.
Общие сведения
Микроцефалия (микрокефалия) – тяжелый порок развития ЦНС, в основе которого лежит уменьшение массы головного мозга и уменьшение окружности черепа более чем на два-три сигмальных отклонения по сравнению со средними половозрастными показателями. Различные формы микроцефалии встречаются с частотой 1 случай на 10000 детей, в равных соотношениях среди мальчиков и девочек. Микроцефалия является причиной олигофрении в 10% наблюдений. При рождении окружность головы у ребенка с микроцефалией, как правило, не превышает 25-27 см (при норме - 35-37 см), а масса головного мозга – 250 г (в норме около 400 г).
Причины микроцефалии
С учетом времени и причин возникновения в педиатрии и детской неврологии выделяют первичную (наследственную, истинную) и вторичную (синдромальную и эмбриопатическую) микроцефалию. Первичная микроцефалия является компонентом наследственных болезней с аутосомно-рецессивным и рецессивным, сцепленным с полом типами наследования (синдром Джакомини, синдром Пейна). На долю истинной микроцефалии приходится 7-34% от всех форм патологии.
Вторичная микроцефалия отмечается при хромосомных аберрациях, наследственных энзимопатиях (фенилкетонурии), патологии беременности и родов. Синдромальная микроцефалия встречается более чем при 125 хромосомных аномалиях, наиболее частыми из которых являются болезнь Дауна (трисомия по 21 хромосоме), синдром Эдвардса (трисомия по 18 хромосоме), синдром Патау (трисомия по 13 хромосоме), синдром «кошачьего крика» (моносомия 5р) и др.
Вторичная эмбриопатическая микроцефалия обусловлена воздействием на плод тератогенных факторов и может являться следствием внутриутробных инфекций (краснухи, цитомегаловирусного энцефалита, герпеса, токсоплазмоза) и интоксикаций (алкогольной, наркотической, профессиональной), радиационного влияния, гипоксии, внутричерепных родовых травм, метаболических нарушений, гормональных заболеваний матери (сахарного диабета, тиреотоксикоза).
Микроцефалия у детей часто сочетается с другими аномалиями: расщелинами губы и неба («заячьей губой» и «волчьей пастью»), несовершенным остеогенезом, врожденной катарактой, пигментным ретинитом, первичной кардиомиопатией, лимфедемой, врожденными пороками сердца и легких, гипоплазией почек, что в значительной мере отягощает прогноз.
Патоморфологическое исследование головного мозга при микроцефалии выявляет уменьшение его массы свыше 25% от нормы, недоразвитие больших полушарий, особенно лобных отделов. При микроцефалии могут иметь место явления микро- или макрогирии (аномально узких либо широких извилин), лиссенцефалии или агирии (сглаженности либо отсутствия извилин), порэнцефалии (наличия патологических кистозных полостей в ткани мозга); агенезия мозолистого тела, расширение ликворных пространств, умеренная гидроцефалия, нарушения миелинизации.
Симптомы микроцефалии
Объем черепа у ребенка с микроцефалией уменьшен уже при рождении, в дальнейшем его развитие заметно отстает от возрастной нормы. Отмечается преобладание лицевого черепа над мозговым. Типичный внешний вид больного с микроцефалией характеризуется узким и скошенным лбом, выступающими надбровными дугами, большими ушами. Большой родничок и черепные швы закрываются уже в первые месяцы жизни. В дальнейшем больные с микроцефалией обычно отстают в массе и росте (вплоть до карликовости), имеют диспропорциональное телосложение, узкое высокое (готическое) небо, большие редкие зубы.
Неврологические нарушения при микроцефалии могут включать мышечную дистонию, спастические парезы, атаксию, судороги, косоглазие. Часто дети с микроцефалией могут страдать эпилепсией и детским церебральным параличом. Дети с микроцефалией поздно начинают держать головку, сидеть, ползать, ходить. Отмечается грубая задержка речевого развития, нечеткость артикуляции, резкая ограниченность словарного запаса, нарушение понимания обращенной речи.
Степень интеллектуальных нарушений у ребенка с микроцефалией может варьировать от дебильности до идиотии. При нерезко выраженной умственной отсталости больные с микроцефалией могут быть обучаемы, способны к самообслуживанию и выполнению несложных поручений. Однако в большинстве случаев дети с микроцефалией требуют ухода, контроля и надзора со стороны взрослых.
По особенностям темперамента дети с микроцефалией могут быть отнесены к торпидной или эретической группе. В первом случае детям свойственна малоподвижность, вялость, безучастность к окружающему, пассивно-подражательная деятельность; во втором случае – гиперактивность, суетливость, подвижность, неустойчивое внимание. Эмоциональная сфера у больных с микроцефалией остается относительно сохранной: дети приветливы, добродушны; реже - эмоционально неустойчивы и склонны к аффективным вспышкам.
Диагностика микроцефалии
Пренатальная диагностика микроцефалии основана на сравнении биометрических параметров плода, получаемых в процессе динамического ультразвукового наблюдения. Однако чувствительность акушерского УЗИ в диагностике микроцефалии составляет всего 67%, а сам порок выявляется только после 27-30 недель беременности. Поэтому при подозрении на микроцефалию, связанную с хромосомной или генетической патологией, УЗИ-скрининг всегда должен дополняться инвазивной пренатальной диагностикой (биопсией хориона, амниоцентезом или кордоцентезом) и кариотипированием плода.
После рождения диагноз микроцефалии подтверждается на основании визуального осмотра новорожденного: уменьшения окружности головы более чем на 2SD-3SD от средней нормы, диспропорции лицевой и мозговой частей черепа. Дети с микроцефалией должны быть проконсультированы генетиком на предмет выявления наследственных заболеваний.
Для определения степени и прогноза микроцефалии важно проведение полного инструментального неврологического обследования: нейросонографии, ЭЭГ, ЭхоЭГ, КТ и МРТ головного мозга. Рентгенография черепа позволяет дифференцировать микроцефалию от краниосиностоза.
Лечение микроцефалии
Патогенетического лечения микроцефалии не существует, поэтому медицинская помощь в основном сводится к симптоматической поддержке больных. На регулярной основе показано проведение медикаментозных курсов, улучшающих обменные процессы в мозговой ткани (пирацетам, пиритинол, витаминные комплексы), по показаниям - противосудорожные и седативные препараты. В рамках реабилитационных мероприятий детям с микроцефалией необходимы занятия лечебной физкультурой, массаж, трудотерапия.
Дети с микроцефалией нуждаются в диспансерном наблюдении педиатра и детского невролога, ежемесячной антропометрии. Воспитание и обучение детей с микроцефалией проводится специалистами дефектологами; коррекция системного недоразвития речи - логопедами. Реабилитационные мероприятия направлены на максимальную адаптацию и социализацию детей с микроцефалией.
Прогноз и профилактика микроцефалии
Прогноз в отношении продолжительности жизни и социализации вариабелен. Некоторые дети способны к обучению в коррекционной школе, овладению элементарными навыками самообслуживания. В целом прогноз при микроцефалии неблагоприятен: продолжительность жизни таких пациентов снижена, большинство из них пожизненно находятся в специнтернатах для умственно-отсталых.
Профилактика микроцефалии у детей предусматривает тщательное планирование беременности, обследование на инфекции (TORCH-комплекс, ПЦР), антенатальную охрану плода. Раннее внутриутробное выявление микроцефалии является основанием для решения вопроса об искусственном прерывании беременности. Медико-генетическое консультирование семей, имеющих детей с микроцефалией, необходимо для оценки потенциального риска при последующих беременностях.
Микроцефалия
Микроцефалия — это неонатальная мальформация, при которой голова ребенка намного меньше, чем у других детей того же возраста и пола. В сочетании с ненадлежащим развитием мозга у детей с микроцефалией могут развиваться нарушения развития. Степень тяжести микроцефалии варьируется от легкой до тяжелой.
Масштабы проблемы
Микроцефалия является редким состоянием. По оценкам, распространенность микроцефалии значительно варьируется из-за разных определений и в зависимости от целевых популяций. Ученые изучают потенциальную, хотя и не доказанную, связь между ростом числа случаев микроцефалии и вирусной инфекцией Зика.
Диагностика
Микроцефалию можно иногда диагностировать с помощью ультразвукового исследования плода. Наиболее подходящим периодом для диагностики является конец второго триместра (около 28 недель) или третий триместр беременности.
Необходимо измерять окружность головы новорожденных, как минимум, через 24 часа после родов и сопоставлять данные со стандартными показателями ВОЗ в области развития детей. Результаты интерпретируются с учетом гестационного возраста ребенка, а также его роста и веса. При наличии подозрений ребенка направляют на осмотр к педиатру и на сканирование мозга, измеряют окружность его головы раз в месяц в раннем грудном возрасте и сопоставляют полученные данные со стандартными показателями. Врачи также должны проводить тесты на известные причины микроцефалии.
Причины микроцефалии
У микроцефалии есть много потенциальных причин, но часто причина остается неизвестной. Наиболее распространенные причины включают:
- внутриутробные инфекции: токсоплазмоз (вызываемый паразитом, обнаруживаемым в мясе, которое не прошло надлежащую тепловую обработку), краснуха, герпес, сифилис, цитомегаловирус и ВИЧ;
- воздействие токсических химических веществ: воздействие на мать тяжелых металлов, таких как мышьяк и ртуть, алкоголя, радиации и курения;
- генетические патологии, такие как синдром Дауна; и
- тяжелая недостаточность питания во время внутриутробного развития.
Признаки и симптомы
У многих детей, рожденных с микроцефалией, при рождении могут отсутствовать другие симптомы, но позже могут развиваться эпилепсия, церебральный паралич, нарушения обучаемости, потеря слуха и проблемы со зрением. Некоторые дети с микроцефалией развиваются совершенно нормально.
Лечение и уход
Специального лечения микроцефалии нет. Для оценки состояния и лечения новорожденных и детей с микроцефалией необходима многопрофильная группа специалистов. Раннее проведение мероприятий по стимулированию и игровых программ может оказывать положительное воздействие на развитие. Семейное консультирование и поддержка родителей также очень важны.
Деятельность ВОЗ
С середины 2015 года ВОЗ тесно сотрудничает со странами Америки в проведении расследований и принятии ответных мер в связи со вспышкой болезни.
В Стратегической программе ответных мер и плане совместных действий изложены шаги, предпринимаемые ВОЗ и партнерами в связи с вирусом Зика и потенциальными осложнениями:
Микроцефалия
Микроцефалия – порок развития ЦНС, характеризующийся уменьшением массы головного мозга и окружности черепа.
Микроцефалия является причиной умственной отсталости и неврологических отклонений. Встречается с одинаковой частотой как среди мальчиков, так и среди девочек. Считается, что на каждые 10000 новорожденных приходится 1 ребенок с микроцефалией. При микроцефалии обнаруживается уменьшение массы головного мозга (в норме составляет приблизительно 400 гр.), окружность головы новорожденного уменьшена до 25 – 27 см (при норме – 35 – 37 см).
Точную причину образования патологии установить достаточно трудно. Возможными факторами являются:
- алкогольная, наркотическая или никотиновая зависимости матери, приводящие к интоксикации плода;
- инфекционные заболевания матери во время беременности (например, токсоплазмоз);
- серьезное токсическое отравление матери во время беременности;
- воздействие радиационного излучения;
- прием токсических доз некоторых лекарственных средств (например, некоторые антибиотики, обладающие тератогенным эффектом);
- различные генетические поломки.
К сожалению, эффективных способов терапии микроцефалии не существует. Основное лечение направлено на уменьшение выраженности симптомов патологии. Для этого применяют ноотропные, седативные, противосудорожные и противоотечные средства, лечебную гимнастику, мануальную терапию. Методами воспитания, обучения и развития личности ребенка вырабатывают элементарные способности самообслуживания и двигательные навыки. Часто дети с подобным диагнозом посещают специальный интернат. Исход заболевания неблагоприятный, патология не подлежит полному излечению. В среднем продолжительность жизни ребенка составляет 15 лет, в редких случаях больные доживают до 30 лет.
Симптомы

Главным симптомом, указывающим на микроцефалию, является уменьшение объема черепа, в том числе преобладание лицевого черепа над мозговым. Также отмечается отставание в массе и росте от возрастных норм, диспропорциональность телосложения.
Учитывая, что головной мозг при микроцефалии не успевает завершить свое развитие, наблюдается отставание ребенка в интеллектуальном и физическом развитии. Поэтому с опозданием формируется речь, артикуляция отличается своей нечеткостью, словарный запас скудный, зачастую наблюдается нарушение понимания обращенной речи. Такие дети поздно начинают держать голову, ползать, сидеть, ходить. Также выявляются спастические парезы, мышечная дистония, атаксия. Часто дети с микроцефалией страдают эпилепсией и детским церебральным параличом. Степень нарушения интеллекта варьирует, при незначительной выраженности умственной отсталости дети с микроцефалией поддаются обучению. Они приспосабливаются к самообслуживанию, а также способны выполнять несложные действия. Но, к сожалению, в большинстве случаев такие люди не способны даже к самообслуживанию, поэтому требуется уход и контроль со стороны взрослых. Такие дети обычно малоподвижны и безучастны к окружающей среде, но при этом они добродушны и приветливы. Реже они отличаются гиперактивным поведением, которое сопровождается вспышками агрессии.
Микроцефалия часто сочетается с другими аномалиями: расщелиной губы и неба, врожденной катарактой, пигментным ретинитом, врожденными пороками сердца и легких, гипоплазией почек. Такие тяжелые аномалии со стороны различным систем органов значительно осложняют состояние человека с микроцефалией, поэтому требуется одновременная медицинская помощь специалистов из разных областей.
Диагностика
Во время беременности каждая женщина обязана проходить УЗИ 1 раз в каждый триместр, при необходимости количество УЗИ увеличивается. Однако микроцефалия на УЗИ обнаруживается на достаточно позднем сроке (25 – 27 недель). Поэтому при подозрении на микроцефалию, связанную с хромосомной или генетической патологией, дополнительно выполняется инвазивная пренатальная диагностика (биопсия хориона, плацентарная биопсия, амниоцентез (биопсия околоплодных вод), кордоцентез (пункция сосудов пуповины, производимая через переднюю брюшную стенку и под контролем УЗИ)). После забора материал отправляется в генетическую лабораторию. Так как инвазивные вмешательства сопряжены с риском для плода, производятся они лишь при согласии женщины и обязательно в условиях стационара, чтобы была возможность наблюдения за состоянием женщины и плода после произведенной процедуры.
При рождении ребенка с микроцефалией обращается внимание на уменьшенный объем черепа, который в дальнейшем не компенсируется, а все также отстает от возрастной нормы. Кроме того, выделяют характерные внешние признаки: преобладание лицевого черепа над мозговым, узкий и скошенный лоб, выступающие надбровные дуги, большие уши, большие редкие зубы, готическое небо, диспропорциональное телосложение. После рождения оценивается скорость закрытия большого родничка. При микроцефалии большой родничок и черепные швы закрываются в течение первого месяца жизни. Для подтверждения микроцефалии в 1й месяц жизни ребенка выполняется нейросонография. Данное ультразвуковое сканирование головного мозга ребенка выполняется через открытый большой родничок. Выявление у ребенка микроцефалии зачастую указывает на наличие генетической патологии, поэтому в диагностических целях назначается консультация генетика.
Для оценки функционального состояния головного мозга назначается ЭЭГ. Этот метод исследования позволяет изучить работу головного мозга, основан на регистрации электрических импульсов, исходящих от его отдельных зон и областей. Является одним из основных методов выявления эпилепсии, которая зачастую развивается при микроцефалии. В дальнейшем для определения степени тяжести и прогноза выполняется МРТ головного мозга.
Лечение

Дети с микроцефалией чаще других должны посещать педиатра, так как требуется регулярный контроль за набором массы тела и увеличением роста ребенка. Также необходимы периодические осмотры неврологом, который производит коррекцию лечения в зависимости от текущего состояния здоровья ребенка. Кроме того, учитывая запоздалое формирование речи, требуется помощь логопеда, занятия с которым не только помогут в становлении речи, но также улучшат артикуляцию и увеличат словарный запас ребенка. Для борьбы со спастичностью необходимы занятия лечебной физкультурой и массаж. Данные занятия должны быть регулярными, так как от их кратности и качества выполнения зависит успех лечения.
Из лекарственных средств используются препараты, улучшающие обменные процессы в ткани головного мозга. Их действие направлено на временное ослабление симптомов со стороны ЦНС, но, к сожалению, они не способны избавить от существующей проблемы полностью. Так как микроцефалия часто сопровождается эпилепсией, назначаются противосудорожные средства. При необходимости используются седативные препараты, благоприятно воздействующие на состояние нервной системы.
Лекарства
Основные лекарственные средства, назначаемые при микроцефалии, направлены на улучшение обменных процессов в головном мозге. За счет этого достигается ослабление симптомов, что приводит к временному улучшению состояния ребенка.
Пирацетам – ноотропное лекарственное средство, благоприятно воздействующее на кровообращение и обменные процессы головного мозга. По данным исследователей препарат способен оказывать следующие эффекты:
- стимулирует интеллектуальную активность;
- улучшает память;
- облегчает процесс обучения;
- повышает умственную работоспособность.
Препарат достаточно хорошо переносится, практически не имеет противопоказаний к применению. Из побочных эффектов, возникающих на фоне приема препарата, выделяют: диспептические симптомы, общую слабость, головокружение, беспокойство, расстройство сна.
Громецин (глицин) также относится к ноотропным средствам. Способен повышать умственную работоспособность, память, концентрацию внимания, нормализует настроение, регулирует незначительное нарушение сна. Побочные эффекты на фоне приема препарата возникают редко. Чаще связаны с индивидуальной непереносимостью и проявляются в виде высыпаний на коже, зуда.
Церебролизин способствует повышению скорости проникновения глюкозы при нарушении гематоэнцефалического барьера, оказывая влияние на уровень ее потребления в поврежденных отделах головного мозга. Кроме того, данный препарат оказывает положительный эффект при нарушении окислительных процессов в структуре обмена веществ головного мозга, снижает уровень церебральной концентрации молочной кислоты. Это приводит к улучшению работы головного мозга.
Также назначаются витамины группы В (неуробекс, боривит) для улучшения проведения нервного импульса.
При микроцефалии часто развивается эпилепсия, поэтому незаменимым компонентом терапии является назначение противосудорожных средств. Подбор конкретного препарата из данной группы, дозировка и кратность приема производится индивидуально каждому пациенту. При этом учитывается тип судорожных припадков, частота их возникновения и чувствительность к медикаментозному лечению. Наиболее подходящий препарат, назначенный квалифицированным неврологом, принимается пациентом постоянно. Это необходимо для предотвращения развития судорожного припадка, во время которого ребенок может получить травмы. Если приступ эпилепсии все же случился, немедленно вводится диазепам. Этот препарат обладает многими терапевтическими действиями, в том числе используется для купирования судорог. Важно понимать, что в качестве поддерживающей терапии данный препарат ни в коем случае не используется, так как существует риск развития привыкания, а также других нежелательных эффектов.
Народные средства

Средства народной медицины используются как дополнительные меры в лечении микроцефалии. Например, всем известный черный шоколад является не только любимым лакомством детей и взрослых, но также, благодаря своему богатству флаванолами, оказывает благоприятное воздействие на умственные способности и настроение человека. Действие реализуется посредством взаимодействия молекул антиоксидантов, стимулирующих перфузию головного мозга.
В китайской медицине с давних времен известно действие женшеня. Это удивительное растение воздействует практически на все процессы мозговой деятельности. Его принимают для улучшения краткосрочной памяти, улучшения внимания, повышения настроения, а также для снижения усталости.
Существует еще одно уникальное растение – родиола розовая. Благодаря исследованиям стало известно, что данное растение способно усиливать работоспособность за счет повышения порога умственного утомления и усталости, вызванной стрессом. Кроме того, выявлены такие действия, как усиление ассоциативного мышления, улучшение краткосрочной памяти, повышение способности к концентрации на различных предметах и явлениях.
В рационе питания должны присутствовать продукты, содержащие достаточное количество омега-3 жирных кислот (грецкие орехи, семена льня, мясо травоядных животных, представители бобовых культур). Кроме того, в аптеке можно приобрести рыбий жир в капсулах, богатый омега-3 жирными кислотами. Данное вещество считается абсолютно заслуженно полезным, так как усиление умственной деятельности наблюдается не только у людей со сниженной интеллектуальной способностью, но также многочисленные исследования привели к выводу, что и у здоровых людей отмечается повышение умственной деятельности. Также следует отдавать предпочтение сложным углеводам при выборе между простыми и сложными. Главным источником сложных углеводов являются крупы. Причем стоит отметить, что речь идет о цельнозерновых продуктах, а не о продуктах их переработки. То есть рекомендуется использовать овес, гречку, пшеницу, коричневый рис. А вот овсяные, гречневые хлопья, манную кашу можно исключить из рациона. Также особое внимание уделяется количеству овощей, съеденному за день. Практически все овоще являются источником сложных углеводов. Следует помнить, что для сохранения полезных свойств овощей, рекомендуется употреблять их в сыром виде.
Информация носит справочный характер и не является руководством к действию. Не занимайтесь самолечением. При первых симптомах заболевания обратитесь к врачу.
Микроцефалия на МРТ
СПбГБУЗ «Детская городская больница №1»;
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
СПбГБУЗ «Детская городская больница №1»;
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
СПбГБУЗ «Детская городская больница №1»
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
Резистентная эпилепсия у ребенка с микроцефально-капиллярным мальформационным синдромом
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2020;120(8): 110‑116
СПбГБУЗ «Детская городская больница №1»;
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
Резистентные к лечению эпилептические энцефалопатии в детском возрасте в большинстве случаев обусловлены генетическими поломками или врожденными аномалиями развития головного мозга. В литературе описан редкий микроцефально-капиллярный мальформационный синдром (Microcephaly-capillary malformation, MIC-CAP), проявляющийся с первого месяца жизни ранним началом резистентной к терапии эпилепсии, тяжелой прогрессирующей микроцефалией, спастическим тетрапарезом, грубой задержкой психомоторного развития, множественными малыми капиллярными ангиомами на туловище и недоразвитием пальцев. У мальчика был диагностирован ранее не описанный вариант нуклеотидной последовательности во 2-м экзоне гена STAMBP chr2:74058171rs781694797 188A>G в гомозиготном состоянии, приводящий к замене аминокислоты p.Tyr63Cys в 63-й позиции белка. Данный вид мутации chr2:74058171rs781694797 188A>G также был выявлен у отца и матери в гетерозиготном состоянии. Данный вариант рассматривается как патогенный, имеющий отношение к фенотипу пациента. В статье приведен обзор литературы с описаниями данного синдрома и собственное наблюдение.
СПбГБУЗ «Детская городская больница №1»;
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
СПбГБУЗ «Детская городская больница №1»;
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
СПбГБУЗ «Детская городская больница №1»
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
Дата принятия в печать:
Микроцефалией традиционно считается уменьшение размера головы на 2 стандартных отклонения или более [1]. Тяжелая микроцефалия — уменьшение окружности головы на более чем 3 стандартных отклонения [1]. По происхождению различают врожденную и постнатальную формы микроцефалии. У большинства больных с врожденной микроцефалией имеется уменьшение числа и уплощение извилин коры; данный паттерн носит название «микроцефалия с обедненным рисунком коры» [1, 2]. Первичная микроцефалия может развиться как изолировано, так и в сочетании с мальформацией коры, недоразвитием мозжечка, перивентрикулярной узловой гетеротопией или другими аномалиями развития мозга [1—3].
Последние исследования показали, что многие пороки развития мозга являются результатом мутаций генов, кодирующих активность внутриклеточного сигнального пути PI3K/AKT/mTOR, центральными компонентами которого являются ферменты фосфоинозитид-3-киназа (PI3K), протеинкиназы AKT и mTOR. Это один из универсальных сигнальных путей, характерных для большинства клеток человека, регулирующий рост, пролиферацию, метаболизм клеток и апоптоз. В пределах ЦНС активация PIK3CA или AKT3 приводит к гиперактивации пути mTOR, что вызывает усиленный перенос РНК и в конечном счете усиливает нейрональную пролиферацию с производством дизморфических нейронов. Клиническая картина значительно варьирует от пациента к пациенту и может включать комбинации полимикрогирии, пахигирии, лиссэнцефалии. Вместе с тем известно, что только 8—30% клеток (нейроны и глия) могут содержать мутированную ДНК, вызывающую развитие диффузной и очаговой кортикальной дисплазии. В этой связи значительное число дисплазий мозга может не выявляться при секвенировании генома.
Впервые данные о синдроме MIC-CAP (Microcephaly-Cutaneous Capillary Malformation Syndrome) были опубликованы M. Carter в 2011 г., им были описаны 2 случая рождения детей с тяжелой первичной микроцефалией, тетрапарезом, ранней младенческой резистентной к лечению эпилепсией и распространенными капиллярными ангиомами, локализованными на туловище [4]. Синдром MIC-CAP включен в базу данных OMIM: 614261. В литературе описаны основные и дополнительные клинические признаки MIC-CAP: лицевые дизморфии, гипоплазия дистальных фаланг, негрубые врожденные дефекты клапанов или перегородок сердца, тугоухость, пузырно-мочеточниковый рефлюкс [4—7] (см. таблицу).
Таблица. Признаки MIC-CAP, по данным литературы
Окружность головы при рождении, центили
Окружность головы при последнем осмотре, центили
Гипоплазия дистальных фаланг кистей или стоп
Аномалия урогенитального тракта
Резистентная неонатальная эпилепсия
Выраженная задержка развития
Малая масса тела к сроку гестации
Микроцефалия с упрощенным рисунком коры
Расширение субарахноидального пространства
Уменьшение размера гиппокампов
Среди краниофациальных стигм дизэмбриогенеза при MIC-CAP описывают круглое лицо, гипертелоризм, короткую спинку носа, низко посаженные ушные раковины, короткую шею, эпикант, птоз, опущенные уголки рта [5]. Большинство случаев имеют клинические проявления с рождения, вместе с тем M. Pavlović описал семейный случай развития заболевания с поздним дебютом в 7 лет и 12 лет. У сибсов отмечались аутистические черты, врожденный гипотиреоз [8].
Патоморфология
Маленькие размеры головного мозга: у одного из умерших детей в возрасте 12 мес размер полушарий соответствовал мозгу новорожденного [5]. У большинства больных выявлены: диффузная кортикальная атрофия, истончение мозолистого тела, редукция белого вещества в области лучистого венца и гиппокампов, гипоплазия ножек мозга и пирамидных трактов, распространенный глиоз зрительных нервов, латеральной петли, зрительной коры, субкортикального белого вещества [5].
Генетика
MIC-CAP имеет аутосомно-рецессивный тип наследования и является результатом гомозиготной мутации в гене STAMBP, локализованной на коротком плече хромосомы 2p13 [4, 5]. При секвенировании описаны различные патогенные мутации в гене STAMBP: p.Arg424Ter [c.1270C>T], p.Phe100Tyr [c.299T>A], p.Arg38Cys [c.112C>T] [4, 5]. В 11 случаях описаны поломки гена в компаунд-гетерозиготном состоянии, в 2 случаях — в гомозиготном состоянии, у 1 пациента — дисомия от 1 родителя. Выявлено 6 миссенс-мутаций, 2 нонсенс-мутации, 3 нарушения рамки считывания, 4 мутации в интроне, приводящие к альтернативному сплайсингу [4]. N. Demikova и соавт. описали характерные фенотипические и нейровизуализационные признаки MIC-CAP у 6-месячной девочки в результате мутации в гене STAMBPc.273delA в гетерозиготном состоянии и интронную замену [c.204-5 C>G] также в гетерозиготном состоянии [5]. Несмотря на то что данный вид мутации не был ранее описан в международной базе данных HGMD и базе данных полиморфизмов dbSN, данный вариант был расценен как патогенный и имеющий отношение к фенотипу пациента [5]. Все это свидетельствует о клиническом полиморфизме MIC-CAP. Носительство мутаций фенотипически асимптомно, поэтому оптимальное время для определения генетического риска и статуса носителя — до наступления беременности [4, 6, 7]. Так как вполне вероятно, что методология тестирования и наше понимание генов и аллельных вариантов болезни в будущем изменятся, следует использовать банковское хранение ДНК у больных с MIC-CAP.
Ген STAMBP кодирует 424-аминокислоты, входящие в состав убиквитиновых ферментов, необходимых для осуществления функции рецепторного аппарата эндоцитов и их дифференцировки [9]. В исследованиях по клеточному метаболизму показано, что РНК-опосредованное уменьшение белка STAMBP в лимфоцитах крови приводит к накоплению агрегатов конъюгированных убиквитинов, что вызывает ослабление активации сигнальных путей передачи сигнала RAS-MAPK и PI3K-AKT-mTOR между клетками [10]. Данный сигнальный путь имеет важное значение в генезе капиллярных или иных новообразований и активации апоптоза, что определяет прогрессирующие нейрональные потери [11, 12].
Прогноз
Продолжительность жизни этих пациентов мало изучена и определяется тяжестью неврологических расстройств. Самый большой возраст ребенка с MIC-CAP, по опубликованным данным, составил 9 лет (данные 2011 г.). В младенчестве умерли 3 детей. Причиной смерти у 1 из них явилось развитие острого панкреатита на фоне лечения вальпроевой кислотой, у 2 других — токсико-септические состояния [4—8].
Описаны случаи использования альтернативных методов лечения (кетогенной диеты) как альтернативного вида контроля над эпилептическими приступами. Эпилептическая активность при MIC-CAP может спонтанно уменьшаться после 3 лет, что позволяет не использовать хирургические виды лечения [6].
Клиническое наблюдение
Мальчик рожден у 30-летней фенотипически здоровой женщины, от второй беременности, протекавшей с острой респираторной вирусной инфекцией на ранних сроках, протеинурией, анемией в III триместре. Отец здоров. Наследственность по эпилепсии не отягощена. Старшая сестра 6 лет здорова. Роды вторые, на сроке 37/38 нед, экстренное кесарево сечение из-за начавшейся внутриутробной гипоксии плода. Масса тела при рождении — 2280 г, длина — 45 см. У мальчика при рождении выявлена задержка внутриутробного развития. Оценка по шкале Апгар — 7/8 баллов. К груди приложен в 1-е сутки, взял неохотно. На 2-е сутки жизни в тяжелом состоянии переведен в ДГБ №1. Тяжесть состояния была обусловлена неврологической симптоматикой: на осмотр реагировал вяло, взгляд не фиксировал, имела место диффузная мышечная гипотония. Обращало на себя внимание наличие краниофациальной дизморфии: окружность головы — 31,5 см (менее 3 центилей), окружность грудной клетки — 34 см (25 центилей). Большое число краниолицевых дизморфий: круглое лицо, широкая и короткая спинка носа, низко посаженные уши, опущенные уголки рта, гипертелоризм (рис. 1, а, б на цв. вклейке). На туловище определялось множество небольших по размеру, до 0,5 см, капиллярных гемангиом (рис. 1, в на цв. вклейке). Также отмечалось укорочение дистальных фаланг пальцев кистей, неправильный рост 3-го пальца на обеих стопах (рис. 1, г, д на цв.вклейке).
Рис. 1. Больной мальчик.
а — в возрасте 5 дней жизни; б — в возрасте 5 мес. Краниолицевые дизморфии: микроцефалия, круглое лицо, широкая и короткая спинка носа, низко посаженные уши, опущенные уголки рта, гипертелоризм; в — множественные округлые капиллярные гемангиомы; г, д — гипоплазия дистальных фаланг пальцев кистей, неправильный рост 3-го пальца на обеих стопах.
На 6-е сутки жизни развился первый генерализованный эпилептический приступ (ЭП) в виде зажмуриваний, вскидывания рук и тонического напряжения мышц шеи и конечностей, с задержкой дыхания. Далее — серийное течение приступов. С возраста 1 мес жизни — прогрессирование ЭП в виде миоклоний, адверсии головы и взора вправо, клонических генерализованных приступов. Проведены многократные электроэнцефалограммы (ЭЭГ): в возрасте 1 мес на ЭЭГ в фоновой записи доминировала медленноволновая активность тета-диапазона амплитудой 80—100 мкВ, на протяжении всей записи отмечены вспышки острых волн и комплексов «острая — медленная волна» амплитудой до 200 мкВ с последующими периодами депрессии ритма — тенденция к формированию паттерна «вспышка — супрессия» (рис. 2, а). Учитывая клиническую картину приступов и данные ЭЭГ, ребенку выставлен диагноз «Младенческая эпилептическая энцефалопатия. Синдром Отахара». На ЭЭГ в возрасте 2 мес — ухудшение фонового ритма, нарастание индекса эпилептиформной активности, тенденция к формированию гипсаритмии (рис.2, б).
Рис. 2. Тот же больной.
а — ЭЭГ в возрасте 1 мес. Вспышки острых волн, комплексов «острая — медленная волна» с последующими периодами амплитудной депрессии ритма. Тенденция к формированию паттерна «вспышка — супрессия»; б — ЭЭГ в возрасте 2 мес. Медленная тета-дельта активность амплитудой до 300 мкВ с включением комплексов «острая — медленная волна» в лобной, левой височной и левой теменно-затылочной областях. Тенденция к формированию гипсаритмии.
При неврологическом осмотре выявлена выраженная диффузная мышечная гипотония с оживлением сухожильных рефлексов, расходящееся косоглазие, гипомимия. До 3 мес и периодически в старшем возрасте кормление осуществляли через зонд, что определялось тяжестью ЭП. Продуктивный контакт к возрасту 4 мес не достигнут. В возрасте 21 дня жизни проведена МРТ головного мозга, выявлен паттерн «микроцефалия с обедненным рисунком коры» (рис. 3, а—в). При повторной МРТ, в возрасте 2 мес жизни, описан паттерн микрогирии в сильвиевой области с двух сторон и диффузное нарастание атрофических изменений полушарий мозга и мозжечка (рис. 3, г—е), что трактовалось при первом исследовании как физиологическое замедление миелинизации в области заднего бедра внутренней капсулы. С возраста 2 мес дисмиелинизация была расценена как патологическая (рис. 4, а, б).
Рис. 3. Тот же больной.
МРТ головного мозга. Паттерн «микроцефалия с обедненным рисунком коры» в возрасте 21 дня жизни (а, б, в) и 2 мес жизни (г, д, е). Паттерн микрогирии в области сильвиевой области указан стрелкой (д).
Рис. 4. Тот же больной.
МРТ головного мозга в возрасте 21 дня (а) и 2 мес (б). Изменение сигнала в области внутренней капсулы указано стрелкой.
На рентгенограмме лучезапястных суставов в прямой проекции в возрасте 4 мес ядра окостенения запястья не определяются, отмечается «бокаловидная» деформация метафизов обеих костей предплечья с разряжением костной структуры.
В возрасте 4 мес проведено генетическое обследование, диагностирован ранее не описанный вариант нуклеотидной последовательности во 2-м экзоне гена STAMBP chr2:74058171rs781694797 188A>G в гомозиготном состоянии, приводящий к замене аминокислоты p.Tyr63Cys в 63-й позиции белка. По результатам секвенирования по Сэнгеру данный вид мутации chr2:74058171rs781694797 188A>G также был выявлен у отца и матери в гетерозиготном состоянии. Данный вариант рассматривается как патогенный, имеющий отношение к фенотипу пациента.
Рис. 5. Тот же больной.
а — в возрасте 7 мес. ЭЭГ — паттерн гипсаритмии; б — в возрасте 12 мес. ЭЭГ — мультифокальная эпилептическая активность.
К возрасту 1,5 года ребенок развивается с грубой задержкой: самостоятельно не сидит, не переворачивается, отсутствует комплекс оживления и зрительно-моторная координация. Сохраняется эпилептический синдром в виде миоклонических приступов с вовлечением мышц лица. Улучшение состоянии было достигнуто на фоне назначения кеппры, ламиктала, клоназепама. В соматическом статусе — рецидивирующие обструктивные бронхиты. УЗИ почек — без особенностей. Глазное дно — частичная атрофия зрительных нервов. Отмечены повышенная чувствительность к шуму, укороченный сон.
Мальчик рожден от второй беременности у фенотипически здоровой женщины. Роды на сроке 37/38 нед. Мальчик родился с признаками внутриутробной гипотрофии (МТ=2280 г, длина=45 см). С рождения у ребенка отмечены врожденная микроцефалия (менее 3 центилей), краниолицевой дисморфизм, множественные капиллярные гемангиомы на коже туловища, гипоплазия дистальных отделов пальцев рук, неправильный рост 3-го пальца стоп. Соматический статус — без особенностей.
В возрасте 1,5 года достигнут неполный клинический контроль над миоклоническими приступами, сохраняются эпилептические спазмы до 5 раз в день. С рождения у ребенка грубая задержка психомоторного развития: к возрасту 1,5 года имеет место диффузная мышечная гипотония центрального типа, выпрямительные реакции не сформированы, продуктивный контакт отсутствует.
Ребенку был проведен анализ кодирующих последовательностей и генов интронно-экзонных сочленений. Выявлен вариант нуклеотидной последовательности во 2-м экзоне гена STAMBP с188A>G в гомозиготном состоянии, не описанный в базе данных. По результатам секвенирования по Сэнгеру данный вид мутации выявлен у отца и матери в гетерозиготном состоянии. Характерные клинические проявления заболевания позволяют отнести данный случай к тяжелой форме MIC-CAP.
Выводы
При развитии резистентных форм младенческой эпилепсии необходимо провести поиск патогенных мутаций как вероятной причины развития заболевания. Выявленный врожденный порок развития головного мозга (микроцефалия) в описанном нами случае является не единственной причиной заболевания. Согласно описанным в литературе клиническим случаям заболевания, имеет место клинический полиморфизм MIC-CAP. При характерных клинических характеристиках MIC-CAP выявлены новые виды мутации, что способствует обновлению имеющейся базы данных. Опираясь на данные литературы о снижении эпилептизации мозга к возрасту 3 лет, целесообразно продолжить комбинированную противоэпилептическую терапию и воздержаться от хирургического метода лечения.
Клинические синдромы нарушения нейрональной пролиферации — семиотика и современная диагностика
Врожденные пороки развития головного мозга (церебральные эмбриофетопатии, дисгенезии или мальформации) встречаются приблизительно у 1 из 100 новорожденных и представляют одну из серьезных проблем современной медицины, т.к. являются причиной антенатальной и ранней детской смертности в 30% [1,2,4,5]. Выжившие дети с пороками головного мозга в дальнейшем являются инвалидами по таким тяжелым заболеваниям, как органические формы умственной отсталости, детские церебральные параличи, различные формы симптоматической эпилепсии [2,3].

С внедрением в практику современных методов исследования нервной системы, в первую очередь методов нейровизуализации (нейросонографии — НСГ, компьютерной томографии — КТ, магнитно-резонансной томографии — МРТ), появилась возможность не только выявлять различные аномалии развития ЦНС прижизненно, но и антенатально [1,2,4,6].
В структуре процессов нейроонтогенеза одним из наиболее важных этапов формирования головного мозга человека является этап нейрональной пролиферации (2—4 месяц внутриутробного развития). Согласно современным данным, головной мозг представляет собой преобразованные стенки эмбриональных мозговых пузырей, полости которых образуют мозговые желудочки. Внутренняя зона мозговых пузырей (перивентрикулярная область) является «зародышевой» зоной для клеток головного мозга. Суть процессов, происходящих на 2—4 месяце внутриутробного развития, заключается в активном размножении нервных клеток (нейрональной пролиферации) в этой области с последующей их радиальной миграцией и размещением в наружной части (краевой зоне) стенки пузыря (будущей коре головного мозга) [2,4,7]. В результате нарушения процессов нейрональной пролиферации развиваются тяжелые структурные нарушения головного мозга, проявляющиеся уменьшением вещества головного мозга (микроцефалия), увеличением вещества головного мозга (мегалэнцефалия), а также нейрокожные синдромы «факоматозы» — туберозный склероз, энцефалотригеминальный ангиоматоз, нейрофиброматоз [2,4,5,8,9]. Практически все болезни, развившиеся вследствие нарушенной нейрональной пролиферации, сопровождаются тяжелой степенью инвалидизации и социальной дезадаптацией [1,2]. Кроме того, обследование детей с различными нарушениями психоречевого и моторного развития выявило, что врожденные аномалии головного мозга, в том числе и болезни нарушений нейрональной пролиферации, являются причиной этих нарушений в 35—40% случаев [2]. В основе нарушения процессов нейрональной пролиферации лежат наследственные факторы (хромосомные и генетические нарушения, тератогенные причины, а также спорадические формы, в том числе обусловленные спонтанными новыми мутациями генов) [2,7].
Целью настоящего исследования явилось выявление с помощью методов лучевой диагностики (КТ, МРТ) особенностей структурной организации головного мозга при следующих клинических синдромах: врожденной микроцефалии, врожденной макроцефалии и различных факоматозов.
Под нашим наблюдением был 41 ребенок с синдромами «нарушения нейрональной пролиферации» в возрасте от 5 суток до 3 лет. Распределение больных по формам синдромов представлено в таблице 1.
Все пациенты обследованы на клинической базе кафедры неонатологии ФУВ ГОУ ВПО РГМУ им. Н.И. Пирогова в отделении компьютерной и магнитно-резонансной томографии Морозовской ДГКБ.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
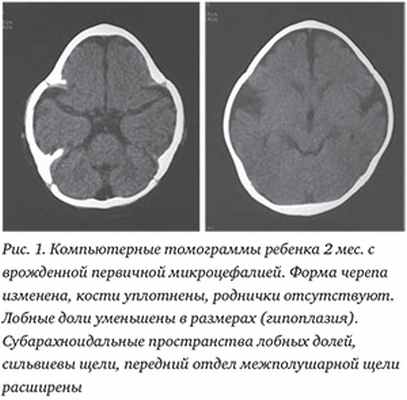
Первичная врожденная микроцефалия характеризовалась малыми размерами головы при рождении, плотными костями, наличием черепных синостозов, малыми размерами черепных родничков или их отсутствием, что согласовалось с данными литературы [2,3,10]. У 4 больных отмечались неонатальные генерализованные клонические судороги. С учетом особенностей строения родничков визуализаци.
Читайте также: