Миоклоническая дистония (миоклонус)
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Миоклоническая дистония или же Синдром миоклонической дистонии это редкое двигательное расстройство, которое вызывает спонтанное сокращение мышц, вызывающее нарушение осанки. О распространенности миоклонической дистонии не сообщалось, однако это заболевание подпадает под действие двигательных расстройств, от которых страдают тысячи людей во всем мире. [1] Миоклоническая дистония возникает в результате мутаций в SGCE ген, кодирующий интегральный мембранный белок, обнаруженный как в нейронах, так и в мышечных волокнах. У страдающих этим заболеванием проявляются симптомы быстрых, резких движений верхних конечностей (миоклонус ), а также искажение ориентации тела из-за одновременной активации мышц-агонистов и антагонистов (дистония ).
Миоклоническая дистония вызвана мутациями потери функции в эпсилон саркогликан ген (SGCE). Заболевание передается доминантно по наследству, однако SGCE является импринтированным геном, [2] так что только по отцовской аллель выражается. Следовательно, дети, страдающие этим заболеванием, наследуют мутацию от отца. Если мутировавший аллель передается по наследству от матери, симптомы у ребенка маловероятны.
Хотя лечения миоклонической дистонии не найдено, для страдающих этим заболеванием доступны варианты лечения. Этанол часто хорошо облегчает симптомы, поэтому синдром также называют «алкогольной дистонией». Алкоголь можно заменить бензодиазепины, Такие как клоназепам, которые работают через тот же механизм. Глубокая стимуляция мозга (DBS) - еще один жизнеспособный вариант, который может облегчить симптомы без нежелательных побочных эффектов лекарств, и оказался успешным при лечении других двигательных расстройств. [3]
Содержание
Признаки и симптомы
Миоклоническая дистония характеризуется двумя основными признаками: миоклонус и дистония. Для большинства людей с миоклонической дистонией миоклонический компонент расстройства часто является основным и наиболее инвалидизирующим признаком по сравнению с компонентом дистонии. Симптомы миоклонической дистонии существенно различаются по степени тяжести.
Миоклонус
Миоклонус характеризуется быстрыми сокращениями, которые влияют на верхнюю часть тела, включая шею, туловище и руки, но могут также влиять на ноги. Эти движения стимулируются различными факторами, включая стресс, шум, кофеин и физические раздражители. Миоклонус можно охарактеризовать по-разному, включая неврологическую основу, мышечную активность и стимулы. Миоклонус может быть положительным или отрицательным; положительный миоклонус возникает в результате кратковременных всплесков мышечной активности, а отрицательный миоклонус возникает при отсутствии какой-либо мышечной активности. Миоклонус обычно классифицируют физиологически для оптимизации лечения. Миоклонус является предшественником миоклонической дистонии и чаще всего начинается в детском или подростковом возрасте. [4] [5]
Миоклонус подразделяется на корковый, подкорковый, периферический и спинномозговой. Кортикальный миоклонус является наиболее распространенным из этих четырех и поражает верхние конечности и лицо. Миоклоническая дистония характеризуется подкорковым происхождением, в частности несегментированным миоклонусом или миоклонусом ствола мозга. Симптомы в рамках этой классификации включают реакцию вздрагивания и ретикулярный рефлекторный миоклонус. Внезапные раздражители, такие как шум или прикосновение к областям вокруг головы или груди, вызывают реакцию испуга, которая распространяется вверх по стволу головного мозга и вниз по спинному мозгу, вызывая резкие движения. Гиперэкплексия это усиленная реакция ствола мозга, при которой пострадавший будет продолжать вызывать тот же ответ на повторяющиеся стимулы. Напротив, ретикулярный рефлекторный миоклонус возникает спонтанно на стимулы, приложенные к дистальным отделам конечностей. Миоклонус позвоночника вызван дефектами в организации или связях позвоночника, а периферический миоклонус имеет симптомы ритмических подергиваний, вызванных нейроном, наиболее распространенным из которых является гемифациальный спазм. [5]
Дистония
Дистония - это реакция на одновременное сокращение мышц-агонистов и антагонистов, которое проявляется как скручивание и искривление, которые влияют на осанку и стойку. Другие симптомы могут включать тремор и мышечные спазмы из-за различных взаимодействий мышц, сокращений и движений. [4] Дистония может быть первичной или вторичной, причем последняя встречается чаще. Первичная дистония или «чистая» дистония имеет только физиологическое происхождение. Вторичная дистония имеет несколько причин: физиологическое, патологическое или неврологическое. [6] [7]
Миоклоническая дистония
Миоклоническая дистония включает в себя быстрые сокращения миоклонии наряду с аномальными позами, классифицируемыми как дистония, а также неврологические и психиатрические проблемы. Это заболевание обычно начинается в детстве с симптомов миоклонии и легкой дистонии, чаще всего цервикальной дистонии или писчая судорога. Симптомы дистонии, как правило, не преувеличиваются в течение болезни и редко являются единственным сопутствующим симптомом, тогда как симптомы миоклонии могут стать более серьезными. Психиатрические проблемы диагностируются клинически на основе вышеупомянутых симптомов и включают депрессию, тревогу, расстройства личности и зависимость. Обсессивно-компульсивное расстройство ассоциирована с миоклонической дистонией, поскольку в различных исследованиях было обнаружено, что у обоих есть общие черты на хромосоме 7. [4]
Неврологические симптомы относительно распространены у пациентов с миоклонической дистонией. Любые неврологические отклонения обычно не проявляются у пациентов в молодом возрасте. Было проведено неврологическое обследование, чтобы определить происхождение этих симптомов, и были выявлены различные части мозга, включая ствол мозга, неокортекс, бледность и таламус. Они вызывают различные эффекты у людей с диагнозом миоклоническая дистония, включая изменения осанки и тремор, и очень редко. слабоумие и атаксия. [4]
Причина
Большинство случаев миоклонической дистонии является результатом мутации в гене эпсилон саркогликана (SGCE ). Этот ген находится на хромосоме 7 с его специфическим цитогенным местоположением 7q21.3. Ген SGCE размером 70 985 п.н. кодирует белок эпсилон (ε) -саркогликан. Пять белков, составляющих семейство саркогликанов, функционируют как интегральные мембранные белки, которые закрепляют цитоскелет ячеек в внеклеточный матрикс. Эпсилон саркогликан - это мембранный белок, который можно найти в печени, легких, почках и селезенке, но наиболее распространен в мышечных и нейронных клетках. Его преобладание как в мышечных волокнах, так и в синапсах нейронов указывает на то, почему симптомы миоклонии и дистонии возникают из-за неправильно функционирующего белка. Рецессивные мутации в других саркогликанах также приводят к мышечным расстройствам, что также подтверждает, что мутации в гене SGCE вызывают миоклоническую дистонию. [8]
Сам саркогликан Эпсилон является частью белок, ассоциированный с дистрофином (DAP) комплекс, который связывает сарколемму мышечных клеток с внеклеточной соединительной тканью. Цель состоит в том, чтобы уменьшить механическое усилие на сарколемма в результате сокращения мышц. Помимо миоклонической дистонии, проблемы, связанные с дисфункциональным комплексом DAP, включают: Мышечная дистрофия Дюшенна. [9]
Считается, что более 65 мутаций гена SGCE вызывают миоклоническую дистонию. Большинство мутаций приводят к образованию усеченного белкового продукта, что приводит к потере функции белка эпсилон-саркогликана. [10] Дисфункциональный белок в конечном итоге перерабатывается клеткой путем разложения, опосредованного протеасома, что приводит к значительной нехватке интегрального мембранного белка как в нейронах, так и в мышечных волокнах.
Мутантный аллель наследуется доминантным образом, то есть мутация может быть унаследована, если один из родителей имеет этот аллель. Тем не мение, геномный импринтинг происходит на аллеле матери, поэтому выражается только аллель отца. [2] [10] Следовательно, наследование мутировавшего отцовского аллеля гена SGCE приведет к экспрессии дисфункционального белка саркогликана эпсилон. Потомство не будет производить мутантный белковый продукт в 95% случаев, когда мать передает мутацию в гене SGCE. [10]
Хотя мутации гена SGCE являются основной причиной миоклонической дистонии, были отдельные случаи, когда отдельные лица и семьи проявляли симптомы, сходные с миоклонической дистонией, но не имели мутаций в этом локусе. Делеции пары оснований гена DYT1, миссенс-мутации в гене DRD2, материнская однопородная дисомия и сцепление хромосомы 18 - все это было связано в редких случаях миоклонической дистонии, когда ген SGCE не затронут. [4]
На сегодняшний день не существует единого универсального метода лечения миоклонической дистонии. Тем не менее, существует несколько методов лечения, которые оказались эффективными для уменьшения симптомов, связанных с синдромом.
Лекарства
Многие лекарства, используемые для лечения миоклонической дистонии, не оказывают значительного воздействия по отдельности, но в сочетании могут воздействовать на различные механизмы мозга, чтобы лучше всего облегчить симптомы. Используемый метод лечения зависит от тяжести симптомов, представленных у человека, и от того, известна ли основная причина синдрома.
Бензодиазепины
Бензодиазепины, такие как клоназепам улучшить тремор, вызванный аспектом миоклонии этого синдрома, аллостерически связываясь с ГАМКА ионотропные рецепторы, вызывающие приток хлорид-ионов, оказывающих тормозящее действие, способное успокоить миоклонические судороги. [4] [11]
Противоэпилептические
Противоэпилептические средства типа вальпроат должен воздействовать на рецепторы ГАМК и управлять ионной проводимостью, чтобы уменьшить тремор и спазмы при миоклонической дистонии. ГАМК-нейроны, которые быстро возбуждаются и воздействуют на моторную кору, блокируются противоэпилептическими средствами в дополнение к изменениям концентраций натрия и кальция, которые могут возбуждать нейрон. Различные противоэпилептические средства различаются по достаточности для контроля ионной проводимости, а также могут вызывать судороги или симптомы миоклонии у некоторых пациентов. [12] Другой использованный агент - зонисамид. [13]
Антихолинергические средства
Антихолинергические препараты, такие как бензатропин облегчить симптомы дистонии, блокируя активность ацетилхолина. Ацетилхолин участвует в патофизиологии дистонии в базальных ганглиях, хотя его точная роль не установлена. Ацетилхолин участвует в дофаминовом и глутаматном путях в базальных ганглиях в дополнение к пресинаптическим мускариновым рецепторам, которые участвуют в моторном контроле. Ацетилхолин обычно сверхактивен у пациентов с дистонией, и блокирование этого нейротрансмиттера уменьшит искривление верхней части тела, но может вызвать побочные эффекты в виде сонливости, спутанности сознания и проблем с памятью у взрослых. [14]
Ботулинический токсин
Ботулинический токсин инъекции также действуют на ацетилхолин, чтобы уменьшить симптомы дистонии. Нейротоксин активен в пресинаптических окончаниях и блокирует экзоцитоз ацетилхолина в синаптическую щель, что снижает мышечную активность. Ботулин также может играть роль в подавлении глутамата и изменении движения мышц. Исследования также показали возможный аксонный транспорт этого нейротоксина, а также его функцию как болеутоляющее, не влияя на сверхактивные движения мышц у пациентов с миоклонической дистонией. [15]
Алкоголь
Также было обнаружено, что употребление алкоголя является эффективным средством для временного ослабления тяжести тремора, связанного с миоклонической дистонией. Алкоголь вызывает усиление передачи ГАМК между интернейронами и клетками Пуркинье. Затем это снижает передачу глутамата в синапсах между гранулярными клетками и клетками Пуркинье, что уменьшает мышечные движения. Эта процедура лишь на короткое время снижает силу тремора и не меняет частоту возникновения тремора. Врачи информируют пациентов о рисках, связанных с употреблением алкоголя при миоклонической дистонии из-за высокой предрасположенности к злоупотреблению алкоголем и зависимости. Сам по себе алкоголизм вызывает тремор в руках и дегенерацию клеток Пуркинье и других частей коры головного мозга, противодействуя первоначальному корректирующему действию алкоголя. [16]
Глубокая стимуляция мозга
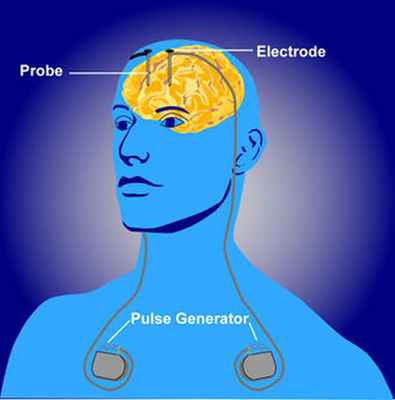
Схема глубокой стимуляции головного мозга пациента. Это распространенный вариант лечения двигательных расстройств, который, как оказалось, помогает облегчить симптомы. [17]
Глубокая стимуляция мозга (DBS) был признан эффективным и безопасным средством лечения пациентов с миоклонической дистонией, у которых тяжелые и изнурительные симптомы резистентны к медикаментозному лечению. Электрическая стимуляция головного мозга является распространенным методом лечения многих двигательных нарушений из-за способности возбуждать или подавлять нейроны в головном мозге. Пациентам с глубокой стимуляцией мозга в мозг вставляют электроды, а затем из внешнего источника посылают электрический сигнал, чтобы вызвать реакцию. Частоту и интенсивность этого сигнала можно изменить, чтобы отслеживать влияние на активность нейронов, используя записи напряжения или нейровизуализацию, например функциональные МРТ. Перемещая электроды в разных областях или изменяя размер или время воздействия раздражителя, можно увидеть различные эффекты на пациента в зависимости от происхождения заболевания. [3]
В других исследованиях изучали влияние DBS на вентро-промежуточное ядро таламус, Vim и Globus pallidus interna, GPi. После глубокой стимуляции мозга с помощью GPi и Vim показатель инвалидности по унифицированной шкале оценки миоклонуса улучшился на 61-66%. Кроме того, оценка по шкале оценки дистомии улучшилась на 45-48%. Хотя не было существенной разницы в улучшении между стимуляцией GPi-Vim и стимуляцией GPi, стимуляция GPi-Vim была значительно более эффективной, чем только глубокая стимуляция мозга Vim. В целом, глубокая стимуляция головного мозга является перспективным средством лечения миоклонической дистонии. [18]
Хотя миоклонус и дистония присутствуют у пациентов с миоклонической дистонией, оптимальное лечение миоклонической дистонии отличается от лечения только миоклонии или дистонии. Миоклонус улучшился значительно больше, чем при дистонии, когда применялась глубокая стимуляция мозга. Кроме того, миоклонус улучшился независимо от того, применялась ли глубокая стимуляция мозга к GPi или Vim. Однако стимуляция GPi была более эффективной для уменьшения симптомов дистонии, чем стимуляция Vim. [17]
Миоклоническая дистония
Миоклоническая дистония – генетически гетерогенное состояние, которое приводит к нарушению работы мышц (миоклоническим гиперкинезам), а также дистонии мускулатуры верхней части тела – шеи, пояса верхних конечностей. Симптомами данного состояния являются резкие мышечные подергивания (рук, шеи, изредка ног), особенно при выполнении тонких движений. Затем присоединяется дистония, которая может проявляться кривошеей и необычной позой больного. Диагностика миоклонической дистонии производится на основании данных настоящего статуса пациента и молекулярно-генетического анализа. Лечение заболевания симптоматическое, включает в себя бензодиазепины и противосудорожные средства, иногда применяют ботулотоксин для устранения спастических нарушений.
Общие сведения
Миоклоническая дистония (миоклонус-дистония, этанол-чувствительная миоклония) – генетическое состояние различной природы, характеризующееся нарушением работы мышц с развитием гиперкинезов и дистонии. Впервые было описано еще в 1940-м году Бенедеком, однако он не смог идентифицировать патологию как отдельный тип миоклонии, это сделали Даубетс и Петерс в 1966-м году. В настоящее время врачи-генетики определили, что это состояние является гетерогенным, то есть за его развитие отвечают мутации различных генов, многие из которых пока не удалось идентифицировать. По этой причине неясен механизм наследования миоклонической дистонии. В большинстве случаев отмечается аутосомно-доминантная передача патологии, при этом имеются указания на наличие материнского импринтинга, при котором дефектный ген, передающийся от матери, не приводит к развитию заболевания. Встречаемость миоклонической дистонии оценивается цифрами 1:500000.
Причины миоклонической дистонии
Достоверно известно, что миоклоническая дистония имеет генетическую природу, при этом встречаются как наследственные семейные случаи, так и спорадические мутации генов. До недавнего времени был известен лишь один ген, мутации которого вызывают развитие данного заболевания – SGCE, локализованный на 7-й хромосоме. В европейской популяции дефекты этого гена встречаются у 20-30% больных миоклонической дистонией. Продуктом его экспрессии является белок саркогликан-эпсилон, относящийся к группе трансмембранных протеинов, встречающийся в скелетных мышцах, миокарде и нейронах ряда структур центральной нервной системы.
Совсем недавно появились указания на то, что у части больных миоклонической дистонией на фоне отсутствия дефектов в SGCE наблюдались мутации гена DRD2, расположенного на 11-й хромосоме. Ген кодирует последовательность одного из рецепторов к дофамину (D2), преобладающего в некоторых базальных ядрах головного мозга. В основном при миоклонической дистонии выявляется миссенс-мутация Val154Ile гена DRD2, приводящая к изменению структуры рецептора. В результате этого меняются многие процессы контроля над мышечным тонусом, что клинически проявляется гиперкинезами и дистонией. Кроме того, при этом заболевании возникают фобии, навязчивые состояния и панические атаки.
Указания на повышенную предрасположенность лиц с мутациями вышеуказанных генов к алкоголизму лишь отчасти имеют под собой основания. Напитки на основе этилового спирта способны ослаблять симптомы миоклонической дистонии, по этой причине больные в ряде случаев употребляют алкоголь в умеренных дозах для облегчения своего состояния, со временем это может стать причиной серьезной зависимости.
Симптомы миоклонической дистонии
При рождении ребенка и в первые годы жизни миоклоническая дистония ничем себя не проявляет, заметить наличие этого состояния не представляется возможным. Первые признаки заболевания возникают в возрасте 15-30 лет, симптоматика патологии нарастает достаточно быстро, но в дальнейшем ее склонность к прогрессированию не выявляется. На начальных этапах миоклонической дистонии развиваются гиперкинезы мышц верхней половины туловища – плечевого пояса, рук, шеи, изредка лица. Иногда подергивания перерастают в судороги, нередко имеющие скручивающий характер и различную частоту возникновения. Гиперкинезы и судороги при миоклонической дистонии редко проявляются в состоянии покоя, в основном их появление связано с мелкими и точными движениями – письмом или рисованием. Мышечные подергивания при этом достаточно резкие, часто описываются больными как «молниеносные».
Еще одним проявлением миоклонической дистонии являются нарушения мышечного тонуса, которые нередко поражают шею, верхние конечности, иногда – мышцы гортани и ног. Наиболее часто отмечается фокальная и спастическая дистония, имеющая асимметричный характер, при развитии на мышцах шеи в подобных случаях возникает кривошея. Как правило, после возникновения дистония не усиливается и не поражает новые участки мышц. В некоторых случаях одним из проявлений миоклонической дистонии может выступать тремор рук. Возможна также дизартрия, обусловленная нарушением функций мышц гортани. Других мышечных симптомов при этом заболевании обычно не определяется.
При миоклонической дистонии нередко выявляются разнообразные расстройства психики: фобии, навязчивые состояния, панические атаки, депрессии. Этанол способен значительно уменьшать выраженность гиперкинезов и дистонии, поэтому многие больные употребляют различные количества алкогольных напитков. При отсутствии врачебного контроля это приводит к стойкой алкогольной зависимости и сопутствующим нарушениям. Поэтому психические симптомы при миоклонической дистонии могут иметь различную природу – первичную, обусловленную нарушением биохимических процессов в нервной системе из-за генетической мутации, и вторичную, вызванную неумеренным употреблением алкоголя.
Диагностика и лечение миоклонической дистонии
Для определения миоклонической дистонии используют данные результатов осмотра больного и молекулярно-генетических анализов. При осмотре выявляют тремор рук, клонические гиперкинезы мышц, плечевого пояса, рук и шеи, которые усиливаются при физической нагрузке или выполнении движений, требующих сложной согласованной работы пучков мышечных волокон (например, рисование). Также может обнаруживаться кривошея, обусловленная спастической дистонией мускулатуры, в более тяжелых случаях определяется необычное положение тела пациента. В анамнезе больного обнаруживаются случаи судорожных припадков и другие неврологические симптомы – все это также косвенно указывает на наличие миоклонической дистонии.
К процессу диагностики миоклонической дистонии зачастую подключают психиатра или нарколога, поскольку при этом состоянии нередко выявляются расстройства психики и проблемы с наркотическими веществами. При обследовании пациента может регистрироваться депрессивное или подавленное состояние, навязчивые идеи и фобии. Панические атаки часто связаны с судорожными припадками, после которых больных миоклонической дистонией начинает одолевать страх смерти, боязнь за свое здоровье или жизнь. Обследование у нарколога иногда обнаруживает признаки алкогольной или (намного реже) бензодиазепиновой зависимости – последняя нередко возникает при неправильном лечении или нарушении схемы приема препаратов.
Молекулярно-генетическая диагностика миоклонической дистонии в настоящее время не получила широкого распространения. В значительной степени это обусловлено тем, что генетические дефекты, приводящие к данному состоянию (мутации генов SGCE и DRD2) достоверно установлены всего лишь примерно для трети случаев заболевания. Однако ряд клиник все же предлагает определение миоклонической дистонии генетическими методами путем прямого секвенирования последовательностей вышеуказанных генов. Пренатальная диагностика необходима в тех случаях, когда миоклонической дистонией страдает отец ребенка – из-за импринтинга передача дефектного гена от матери потомству происходит крайне редко. Дифференциальную диагностику производят с семейной миоклонией (болезнью Унферрихта-Лундборга), синдромом Туретта, болезнью Вильсона, другими первичными и вторичными формами дистонии.
Специфического лечения миоклонической дистонии не существует, применяют симптоматическую терапию. При наличии тремора и выраженных гиперкинезов хорошим эффектом обладают препараты из группы бензодиазепинов, однако назначать их можно только после консультации нарколога – важно, чтобы у больного не было алкогольной или бензодиазепиновой зависимости. Судорожные припадки, возникающие при миоклонической дистонии, устраняются при помощи традиционной противосудорожной терапии. Спастические дистонии мышц и в особенности кривошея могут быть значительно ослаблены инъекциями малых доз ботулинового токсина для частичной химической денервации мышечной ткани. Эффективность перечисленных терапевтических мероприятий неодинакова у различных больных миоклонической дистонией, что обусловлено значительной генетической и фенотипической гетерогенностью данного состояния.
Прогноз и профилактика миоклонической дистонии
Прогноз миоклонической дистонии относительно выживаемости пациентов довольно благоприятный – состояние не имеет склонности к прогрессированию симптомов, не поражает жизненно важные системы и органы. Однако качество жизни больных может в значительной степени снижаться из-за гиперкинезов, которые нередко делают невозможными выполнение тонких движений (например, письмо), дистонической кривошеи и других симптомов заболевания. Кроме того, прогноз миоклонической дистонии ухудшает повышенный риск развития алкогольной зависимости – даже благополучный в социальном плане человек может пристраститься к спиртосодержащим напиткам из-за того, что они облегчают неврологические симптомы. Еще больше повышают риски психоэмоциональные расстройства пациента, которые увеличивают вероятность развития алкогольной зависимости и могут стать причиной серьезного невроза. Специфической профилактики миоклонической дистонии не существует, при возникновении заболевания следует выполнять все предписания врача.
Миоклония
Миоклония – это кратковременное быстрое сокращение мышцы или группы мышц. Диагноз устанавливают на основании клинических проявлений и иногда подтверждают результатами электромиографического исследования. Лечение включает в себя коррекцию обратимых патологических состояний, и при необходимости пероральную симптоматическую терапию.
Классификация миоклонуса
Физиологическая миоклония может наблюдаться во время засыпания и ранних фаз сна (гипнагогическая миоклония). Гипнагогические миоклонии могут быть очаговыми, мультифокальными, сегментарными, или генерализованными (см. ниже), и могут напоминать реакцию испуга. Другой тип физиологических миоклоний - икота (диафрагмальные миоклонии).
Патологические миоклонии могут развиваться при различных заболеваниях, а также при применении лекарственных препаратов (см. таблицу Некоторые причины миоклонуса [Some Causes of Myoclonus] Некоторые причины миоклонуса ). Самыми частыми причинами являются:
Другие причины патологического миоклонуса включают дегенеративные заболевания с поражением базальных ганглиев и некоторые виды деменции.
Выделяют следующие типы амнезий:
По распределению: фокальные, сегментарные (смежные области), мультифокальные (несмежные области) или генерализованные
По месту происхождения: кортикальные, подкорковые, сегментарные или периферические
По клиническим проявлениям: положительные или отрицательные
По своей этиологии: эссенциальные (первичные), приобретенные или идиопатическые
По триггерному фактору: сенсорные или спонтанные
Миоклонус классифицируется по месту возникновения следующим образом:
Кортикальный: кортикальный миоклонус связан с повреждением коры головного мозга или эпилепсией. Световые зрительные раздражители или прикосновения могут спровоцировать миоклонические судороги, которые могут вызвать появление отклонений на электроэнцефалограмме (например, очаговые или генерализованные комплексы пик-волна или множественные эпилептиформные разряды полипик-волна, гигантские соматосенсорные вызванные потенциалы). Миоклонические подергивания могут быть менее явными в состоянии покоя, но усугубляться во время двигательной активности. Этот тип миоклонуса может серьезно ухудшить речь и походку.
Сегментарные и периферические: формы сегментарного или периферического миоклонуса встречаются относительно редко. Сегментарный миоклонус включает спинальный сегментарный и проприоспинальный миоклонус. Спинальный сегментарный миоклонус относится к миоклонусу в спинных мышцах одного или нескольких смежных сегментов спинного мозга. Проприоспинальный миоклонус характеризуется медленно распространяющимися движениями, щадящими лицо, часто внезапными, напоминающими удар, что не характерно для других типов миоклонуса. Нёбный миоклонус, который в настоящее время в основном считается ошибочным обозначением, был классифицирован как тремор неба. Наиболее распространенным периферическим миоклонусом является гемифациальный спазм Гемифациальный спазм Гемифациальный спазм – односторонние безболезненные синхронные сокращения мышц лица вследствие повторяющихся непроизвольных электрических импульсов 7 черепного (лицевого) нерва и/или его двигательного. Прочитайте дополнительные сведения , он возникает в основном из-за сосудистого сдавливания лицевого нерва при его выходе из ствола мозга или при опухолях мосто-мозжечкового угла. Полугемифациальный спазм встречается гораздо реже; он характеризуется односторонними пароксизмальными сокращениями мышц челюсти. Он может быть вызван компрессией моторной ветви тройничного нерва.
Классификация миоклонуса, основанная на месте возникновения, считается наиболее целесообразной при выборе наиболее эффективной терапии.
Клиническая картина пациентов с миоклонусом может быть классифицирована как положительная или отрицательная:
Положительный: у пациентов наблюдается активные подергивания мышц, приводящие к резкому движению.
Негативный: внезапно падает мышечный тонус (биоэлектрическое молчание на электромиографии); когда мышечный тонус теряют опорные (антигравитационные) мышцы, пациент может упасть. К негативному миоклонусу относится астериксис (трепетание рук, которое возникает у пациентов с тяжелой печеночной недостаточностью).
У одного и того же пациента часто могут наблюдаться как позитивный, так и негативный миоклонусы.
Этиология миоклонии может быть эссенциальной (первичной), приобретенной (наиболее распространенной) или идиопатической.
При эссенциальном (первичном) миоклонусе идентифицируемая причина отсутствует и/или предполагается наличие генетических факторов.
Приобретенный миоклонус имеет несколько причин, в том числе многие метаболические нарушения (см. таблицу Некоторые причины миоклонуса Некоторые причины миоклонуса ). Большинство случаев миоклонуса являются приобретенными.
Идиопатический миоклонус является миоклонусом, наличие которого полностью необъяснимо.
Миоклония может иметь триггерный фактор или же он может отсутствовать:
Сенсорно-чувствительный: миоклония вызывается внешним стимулом (например, внезапным шумом, движением, светом, визуальной угрозой), как это происходит, когда человек внезапно напуган (реакция испуга).
Спонтанный: миоклония возникает без триггерного фактора, как это часто бывает, когда причиной является метаболизм.
ИЗУЧЕНИЕ ФЕНОМЕНА КУПИРОВАНИЯ ЭТАНОЛОМ И ТЕРАПЕВТИЧЕСКИХ ДОЗ БЕНЗОДИАЗЕПИНОВ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ СИНДРОМА МИОКЛОНУС ДИСТОНИИ. КЛИНИЧЕСКИЙ СЛУЧАЙ Текст научной статьи по специальности «Клиническая медицина»
Текст научной работы на тему «ИЗУЧЕНИЕ ФЕНОМЕНА КУПИРОВАНИЯ ЭТАНОЛОМ И ТЕРАПЕВТИЧЕСКИХ ДОЗ БЕНЗОДИАЗЕПИНОВ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ СИНДРОМА МИОКЛОНУС ДИСТОНИИ. КЛИНИЧЕСКИЙ СЛУЧАЙ»
Abstracts Nationwide scientific forum of students with international participation «STUDENT SCIENCE - 2021»
изучение феномена купирования этанолом и терапевтических доз бензодиазепинов клинических проявлений синдрома миоклонус-дистонии. клинический случай
Научный руководитель: профессор, д.м.н, Васильев А.Г., ассистент к.м.н Брус Т.В., старший лаборант Пюрвеев С.С Кафедра патологической физиологии с курсом иммунопатологии Санкт-Петербургский государственный педиатрический медицинский университет
ключевые слова: миоклоническая дистония(миоклонус-дистония), бензодиазепины, этанол-зависимая дистония
Актуальность исследования: По данным ВОЗ миоклоническая дистония считается достаточно редким заболеванием, частота выявленных случаевоколо 1:500.000 в Европе, в связи с чем относится к орфанным заболеваниям- мало изученным, и, как правило, сложными в лечении [1]
цель исследования: изучить патогенез миоклоний и дистонии при данном заболевании, наблюдение за больным с данным заболеванием.
Материалы и методы: Клинический случай подтвержденного заболевания, исследования литературы. Клинический случай: пациент (19.09.00), поступление в центр экстрапирамидных расстройств с миоклоническими судорогами и дистонией шейных мышц. Диагноз поставлен врачом-паркинсонологом на основании жалоб, анамнеза жизни и заболевания, объективному и инструментальному обследованию (МРТ головного мозга и шейного отдела позвоночника, игольчатая ЭМГ, ЭЭГ)
Результаты: Рассмотрен клинический случай заболевания: девушка, 20 лет, жалобы на ми-оклонические судороги в нижних конечностях, туловище, голове, боли в шее, слабости мышц верхних конечностей с первой половины 2017 года. Семейный анамнез отягощен: у тети эпилепсия, подобных симптомов в семье до третьего поколения не наблюдалось. Ребенок от первых родов, развитие по возрасту. При объективном осмотре явные миоклонические судороги, дистония шейных мышц, слабость мышц верхней конечности. терапия противосудорожными (вальпроаты, карбамазепин, габапентин, леветирацетам) не дала положительного эффекта. Неврологический статус: высшие мозговые функции сохранены. МРТ шейного отдела позвоночника и головного мозга без патологии. Складывается впечатление о легком тортиколлисе влево. Поля зрения, реакция и точки конвергенции в норме. Нистагма нет. Точки выхода тройничного нерва безболезненны. Симптом орального автоматизма отсутствует. Слух, фонация не нарушены, язык по средней линии. Нет нарушений чувствительности. Рефлексы в норме, отсутствие патологических рефлексов, в позе Ромберга устойчив. менингеальные симптомы отсутствуют. Выписана терапия клоназепамом (0.5 мг по по таблетки три раза в день)[2]. Видна положительная динамика (уменьшение частоты и амплитуды судорог) с побочными эффектами в виде редких панических атак и сонливости.
Выводы: генетическая природа заболевания даёт возможность предположить, что помимо классической терапии бензодиазепинами и стимуляцией глубинных структур подкоркового вещества мозга, возможна генная терапия, как на примере переноса генов с мышечной дистрофией плечевого пояса конечностей типа 2D (LGMD2D), что может полностью купировать симптомы больных.[3]
1. Электронный ресурс: Expert reviewer(s): Dr Christoph KAMM; OrphaNet; Ноябрь 2017
2. Электронный ресурс: Vidal-механизмы действия бензодиазеапинов и этанола
3. Электронный ресурс: National Institute of Arthritis and Musculoskeletal and Skin Diseases
Пилотное исследование эффективности Т2000 при миоклонической дистонии
Фаза II: эффективность и безопасность пролекарства T2000 (1,3-диметоксиметил-5,5-дифенилбарбитуровая кислота) Taro Pharmaceuticals у пациентов с миоклонической дистонией: открытое исследование последовательного повышения дозы
Это пилотное исследование будет оценивать безопасность и эффективность приема T2000 один раз в день при использовании для лечения пациенты с миоклонической дистонией в течение 12 недель.
Миоклоническая дистония (М-Д) - редкое наследственное двигательное расстройство, при котором пациенты испытывают миоклонус - внезапные, короткие, непроизвольные отрывистые движения, часто связанные с дистонией - непроизвольные устойчивые сокращения, вызывающие скручивание или неправильную осанку. Хотя большинство M-D пациенты значимо реагируют на алкоголь, одобренных лекарств от М-Д нет. А в настоящее время для лечения M-D используются различные лекарства, но эти методы лечения работают в небольших доля пациентов и обеспечивает лишь частичное улучшение симптомов; их использование также ограничено побочными эффектами у многих пациентов. T2000 - это разрабатываемый в настоящее время препарат для лечения двигательных нарушений, включая эссенциальный тремор (ЭТ). Хотя T2000 - новый препарат, он принадлежит к классу лекарства, которые использовались в течение многих лет для лечения различных медицинских условия. В предыдущих исследованиях T2000 оказался эффективным в борьбе с симптомами ЭТ. а у некоторых пациентов с тяжелой формой ЭТ тремор значительно улучшился. Как и следовало ожидать от лекарства этого класса, T2000 могут вызывать седативный эффект при высоком уровне в крови, который может наблюдаться когда большие дозы даются пожилым людям. У более молодых пациентов T2000 вызывал только минимальные побочные эффекты даже при приеме в высоких дозах и в течение периодов от нескольких недель до несколько месяцев. Текущее исследование будет оценивать безопасность и эффективность T2000 у пациентов с M-D. Пациенты будут получать дозы T2000, начиная с 200 мг в день и постепенно увеличивая их. в неделю на дополнительные 200 мг в день до максимальной дозы 1000 мг в день. Общая продолжительность лечения составит 12 недель. Симптомы миоклонии и дистонии у пациента, а также общее неврологическое обследование будет контролироваться на протяжении всего исследования. Ответ на T2000 будет определяться путем сравнения степени тяжести миоклонии и дистонии, пока пациенты получают T2000 по сравнению с симптомами, наблюдаемыми без активного лечения.
Тип вмешательства: Препарат, средство, медикамент
Описание: T2000 в дозах от 200 мг в сутки до 1000 мг в сутки
Этикетка Arm Group: 1
Критерии включения: - Пациенты должны соответствовать диагностическим критериям M-D, основанным на следующих критериях: - миоклонус - первичный признак; фокальная или сегментарная дистония любой степени тяжести может также присутствовать - симптомы начались к 20 годам - должен присутствовать семейный уклад - неврологический анамнез не должен наводить на мысль о другом неврологическом состояние - такие исследования, как визуализация, ЭЭГ и тесты на вызванный потенциал, должны быть нормальными - Пациенты будут иметь право на участие в этом исследовании, если у них в настоящее время лечения, не переносят текущие методы лечения или не проходят лечение у пациентов, у которых были объяснены альтернативы лечения. Критерий исключения: - Пациенты адекватно контролируются без побочных эффектов при текущем лечении M-D - Текущее лечение барбитуратами, такими как фенобарбитал или примидон - Беременные пациенты или пациенты, которые могут забеременеть во время исследования. - Пациенты, которым необходимо принимать лекарства, изменяющие метаболизм печени, а также пациенты при заболеваниях печени или нарушениях свертывания крови - Пациенты с судорожными расстройствами - Пациенты с аллергией или реакцией гиперчувствительности на барбитураты или другие родственные лекарства, такие как фенобарбитал или фенитоин - Пациенты со значительными общими медицинскими или клиническими лабораторными отклонениями
Распределение: Нет данных
Модель вмешательства: Назначение одной группы
Первичное назначение: лечение
Маскировка: Нет (открытая этикетка)
This information was retrieved directly from the website clinicaltrials.gov without any changes. If you have any requests to change, remove or update your study details, please contact [email protected] . As soon as a change is implemented on clinicaltrials.gov, this will be updated automatically on our website as well.
Клинические исследования Миоклонус
Заболевания: дистония, Медикаментозная дистония, Болезнь Паркинсона, Эссенциальный тремор, Дискинезии, Миоклонус, Тиковые расстройства, Кривошея, Защемление локтевого нерва, Заболевания височно-нижнечелюстного сустава, Дисфония
Заболевания: Аутоиммунное заболевание, Неврологическое аутоиммунное заболевание, Аутологичный трансплантат Аутоиммунный, Трансплантация рассеянного склероза, Трансплантация стволовых клеток рассеянного склероза, Трансплантация стволовых клеток рассеянного склероза, Синдром жесткого человека, HCT для неврологических аутоиммунных расстройств, ХВДП Трансплантация, Трансплантация миастении гравис, Аутоиммунное заболевание нервной системы, Васкулит центральной нервной системы, Мозжечковая дегенерация, Хроническая воспалительная демиелинизирующая полинейропатия, Миастенический синдром Ламберта Итона, Миастения Гравис, Оптический нейромиелит, Опсоклонус-миоклонус-синдром, Подострый энцефалит Расмуссена
Заболевания: Редкие заболевания, Недиагностированные расстройства, Заболевания неизвестной распространенности, Синдром Корнелии де Ланге, Пренатальная доброкачественная гипофосфатазия, Перинатальная летальная гипофосфатазия, Одонтогипофосфатазия, Взрослая гипофосфатазия, Гипофосфатазия в детском возрасте, Детская гипофосфатазия, Гипофосфатазия, Синдром Кабуки, Синдром Боринга-Опица, Нарколепсия без катаплексии, Нарколепсия-катаплексия, Гиперсонное расстройство, Идиопатическая гиперсомния без продолжительного сна, Идиопатическая гиперсомния с длительным временем сна, Идиопатическая гиперсомния, Синдром Кляйне-Левина, Болезнь Кавасаки, лейомиосаркома, Лейомиосаркома тела матки, Лейомиосаркома шейки матки, Лейомиосаркома тонкой кишки, Приобретенная миастения гравис, Болезнь Аддисона, Гиперакузия (Гиперакузия), Ювенильная миастения гравис, Транзиторная неонатальная миастения гравис, Синдром Вильямса, Болезнь Лайма, Миастения Гравис, Синдром Маринеско-Шегрена (синдром Маринеско-Шегрена), Изолированный синдром Клиппеля-Фейля, Синдром Фрейзера, Синдром Дениса-Драша, Синдром Беквита-Видеманна, Синдром Эмануэля, Изолированная аниридия, Синдром Аксенфельда-Ригера, Аниридия-синдром умственной отсталости, Аниридия - Агенезия почек - Задержка психомоторного развития, Аниридия - Птоз - Умственная отсталость - Семейное ожирение, Аниридия - мозжечковая атаксия - умственная отсталость, Аниридия – отсутствие надколенника, Аниридия, Аномалия Питерса - Катаракта, Аномалия Питерса, Синдром Потоцкого-Шаффера, Синдром Сильвера-Рассела из-за однородительской дисомии хромосомы 11 у матери, Синдром Сильвера-Рассела из-за дефекта импринтинга 11p15, Синдром Сильвера-Рассела из-за микродупликации 11p15, Синдромная аниридия, WAGR-синдром, Синдром Вольфа-Хиршхорна, 4p16.3 Синдром микродупликации, Синдром делеции 4p, синдром не-Вольфа-Хиршхорна, Аутосомно-рецессивный синдром Стиклера, Синдром Стиклера 2 типа, Синдром Стиклера Тип 1, Синдром Стиклера, Муколипидоз 4 типа, Х-сцепленная спиноцеребеллярная атаксия типа 4, Х-сцепленная спиноцеребеллярная атаксия типа 3, Х-сцепленная умственная отсталость - атаксия - апраксия, Х-сцепленная прогрессирующая мозжечковая атаксия, Х-сцепленная непрогрессирующая мозжечковая атаксия, Х-сцепленная мозжечковая атаксия, Атаксия при дефиците витамина B12, Токсическое воздействие Атаксия, Неклассифицированная аутосомно-доминантная спиноцеребеллярная атаксия, Атаксия антител к щитовидной железе, Спорадическая атаксия неизвестной этиологии у взрослых, Спиноцеребеллярная атаксия с глазодвигательной аномалией, Спиноцеребеллярная атаксия с эпилепсией, Спиноцеребеллярная атаксия с аксональной невропатией 2 типа, Спиноцеребеллярная атаксия 8 типа, Спиноцеребеллярная атаксия 7 типа, Спиноцеребеллярная атаксия 6 типа, Спиноцеребеллярная атаксия 5 типа, Спиноцеребеллярная атаксия 4 типа, Спиноцеребеллярная атаксия 37 типа, Спиноцеребеллярная атаксия 36 типа, Спиноцеребеллярная атаксия 35 типа, Спиноцеребеллярная атаксия 34 типа, Спиноцеребеллярная атаксия 32 типа, Спиноцеребеллярная атаксия 31 типа, Спиноцеребеллярная атаксия 30 типа, Спиноцеребеллярная атаксия 3 типа, Спиноцеребеллярная атаксия 29 типа, Спиноцеребеллярная атаксия 28 типа, Спиноцеребеллярная атаксия 27 типа, Спиноцеребеллярная атаксия 26 типа, Спиноцеребеллярная атаксия 25 типа, Спиноцеребеллярная атаксия 23 типа, Спиноцеребеллярная атаксия 22 типа, Спиноцеребеллярная атаксия 21 типа, Спиноцеребеллярная атаксия 20 типа, Спиноцеребеллярная атаксия 2 типа, Спиноцеребеллярная атаксия Тип 19/22, Спиноцеребеллярная атаксия 18 типа, Спиноцеребеллярная атаксия 17 типа, Спиноцеребеллярная атаксия 16 типа, Спиноцеребеллярная атаксия Тип 15/16, Спиноцеребеллярная атаксия 14 типа, Спиноцеребеллярная атаксия 13 типа, Спиноцеребеллярная атаксия 12 типа, Спиноцеребеллярная атаксия 11 типа, Спиноцеребеллярная атаксия 10 типа, Спиноцеребеллярная атаксия типа 1 с аксональной невропатией, Спиноцеребеллярная атаксия типа 1, Спиноцеребеллярная атаксия - неизвестно, Спиноцеребеллярная атаксия – дисморфизм, Непрогрессирующая эпилепсия и/или атаксия с миоклонусом в качестве основного признака, Синдром спастичности-атаксия-аномалии походки, Спастическая атаксия с врожденным миозом, Спастическая атаксия - дистрофия роговицы, Спастическая атаксия, Редкая наследственная атаксия, Редкая атаксия, Синдром рецессивной митохондриальной атаксии, Прогрессирующая эпилепсия и/или атаксия с миоклонусом в качестве основного признака, Атаксия задней колонны — пигментный ретинит, Постинсультная атаксия, Атаксия после травм головы, Поствакцинальная атаксия, Полинейропатия - Потеря слуха - Атаксия - Пигментный ретинит - Катаракта, Мышечная атрофия - Атаксия - Пигментный ретинит - Сахарный диабет, Ненаследственная дегенеративная атаксия, Пароксизмальный дистонический хореатетоз с эпизодической атаксией и спастичностью, Оливопонтоцеребеллярная атрофия - глухота, НАРП-синдром, Миоклонус - мозжечковая атаксия - глухота, Множественная системная атрофия, паркинсонический тип, Множественная системная атрофия мозжечкового типа, Множественная системная атрофия, Синдром Ли, наследуемый по материнской линии, Болезнь Мачадо-Жозефа 3 типа, Болезнь Мачадо-Жозефа Тип 2, Болезнь Мачадо-Жозефа Тип 1, Синдром Ли, Поздняя атаксия с деменцией, Инфекционная или постинфекционная атаксия, ГТР Атаксия, Наследственная эпизодическая атаксия, Глиадин/глютеновая атаксия, Фридрих Атаксия, Хрупкий Х-ассоциированный тремор/синдром атаксии, Семейная пароксизмальная атаксия, Воздействие лекарств Атаксия, Эпизодическая атаксия с невнятной речью, Эпизодическая атаксия неизвестного типа, Эпизодическая атаксия 7 типа, Эпизодическая атаксия 6 типа, Эпизодическая атаксия 5 типа, Эпизодическая атаксия 4 типа, Эпизодическая атаксия 3 типа, Эпизодическая атаксия типа 1, Эпилепсия и/или атаксия с миоклонусом в качестве основного признака, Синдром спастической атаксии-нейропатии с ранним началом, Прогрессирующая нейродегенерация с ранним началом - слепота - атаксия - спастичность, Ранняя мозжечковая атаксия с сохраненными сухожильными рефлексами, Ранняя атаксия с деменцией, Аутосомно-рецессивная медленно прогрессирующая спиноцеребеллярная атаксия в детском возрасте, Дилатационная кардиомиопатия с атаксией, Катаракта - Атаксия - Глухота, Мозжечковая атаксия Кайманова типа, Мозжечковая атаксия с периферической нейропатией, Мозжечковая атаксия - гипогонадизм, Мозжечковая атаксия - эктодермальная дисплазия, Мозжечковая атаксия - Арефлексия - Полая ступня - Атрофия зрительного нерва - Нейросенсорная тугоухость, Атаксия опухоли головного мозга, Брахидактилия - нистагм - мозжечковая атаксия, Доброкачественный пароксизмальный тонический взгляд вверх в детском возрасте с атаксией, Аутосомно-рецессивная синдромальная мозжечковая атаксия, Аутосомно-рецессивная спастическая атаксия с лейкоэнцефалопатией, Аутосомно-рецессивная спастическая атаксия Шарлевуа-Сагенея, Аутосомно-рецессивная спастическая атаксия - атрофия зрительного нерва - дизартрия, Аутосомно-рецессивная спастическая атаксия, Аутосомно-рецессивная метаболическая мозжечковая атаксия, Аутосомно-доминантная спиноцеребеллярная атаксия из-за повторяющихся экспансий, не кодирующих полиглутамин, Аутосомно-рецессивная атаксия, тип Beauce, Аутосомно-рецессивная атаксия из-за дефицита убихинона, Аутосомно-рецессивная атаксия из-за дефицита PEX10, Аутосомно-рецессивная дегенеративная и прогрессирующая мозжечковая атаксия, Аутосомно-рецессивная врожденная мозжечковая атаксия из-за дефицита MGLUR1, Аутосомно-рецессивная врожденная мозжечковая атаксия из-за дефицита GRID2, Аутосомно-рецессивная врожденная мозжечковая атаксия, Аутосомно-рецессивная мозжечковая атаксия-пирамидные признаки-нистагм-окуломоторный синдром апраксии, Аутосомно-рецессивная мозжечковая атаксия-эпилепсия-синдром умственной отсталости из-за дефицита WWOX, Аутосомно-рецессивная мозжечковая атаксия-эпилепсия-синдром умственной отсталости из-за дефицита TUD, Аутосомно-рецессивная мозжечковая атаксия-эпилепсия-синдром умственной отсталости из-за дефицита KIAA0226, Аутосомно-рецессивная мозжечковая атаксия-эпилепсия-синдром умственной отсталости, Аутосомно-рецессивная мозжечковая атаксия с поздним началом спастичности, Аутосомно-рецессивная мозжечковая атаксия из-за дефицита STUB1, Аутосомно-рецессивная мозжечковая атаксия из-за дефекта репарации ДНК, Аутосомно-рецессивная мозжечковая атаксия - саккадическая интрузия, Аутосомно-рецессивная мозжечковая атаксия - задержка психомоторного развития, Аутосомно-рецессивная мозжечковая атаксия - слепота - глухота, Аутосомно-рецессивная мозжечковая атаксия, Аутосомно-доминантная спиноцеребеллярная атаксия, обусловленная полиглутаминовой аномалией, Аутосомно-доминантная спиноцеребеллярная атаксия вследствие точечной мутации, Аутосомно-доминантная спиноцеребеллярная атаксия, обусловленная каналопатией, Аутосомно-доминантная спастическая атаксия 1 типа, Аутосомно-доминантная спастическая атаксия, Аутосомно-доминантная атрофия зрительного нерва, Вариант атаксии-телеангиэктазии, Атаксия-телеангиэктазия, Аутосомно-доминантная мозжечковая атаксия, глухота и нарколепсия, Аутосомно-доминантная мозжечковая атаксия 4 типа, Аутосомно-доминантная мозжечковая атаксия 3 типа, Аутосомно-доминантная мозжечковая атаксия 2 типа, Аутосомно-доминантная мозжечковая атаксия 1 типа, Аутосомно-доминантная мозжечковая атаксия, Расстройство, подобное атаксии-телеангиэктазии, Атаксия при дефиците витамина Е, Атаксия с деменцией, Атаксия - глазодвигательная апраксия 1 типа, Атаксия – другое, Атаксия - Генетический диагноз - Неизвестно, Приобретенная атаксия, Аутосомно-рецессивная мозжечковая атаксия у взрослых, Атаксия, связанная с алкоголем, Множественная эндокринная неоплазия, Множественная эндокринная неоплазия II типа, Множественная эндокринная неоплазия 1 типа, Множественная эндокринная неоплазия 2 типа, Множественная эндокринная неоплазия, тип IV, Множественная эндокринная неоплазия, тип 3, Синдром множественной эндокринной неоплазии (МЭН), Множественная эндокринная неоплазия типа 2В, Множественная эндокринная неоплазия типа 2А, Атипичный гемолитико-уремический синдром, Атипичный ГУС, Синдром Видемана-Штайнера, Анапластическая крупноклеточная лимфома, связанная с грудным имплантатом, Аутоиммунный/воспалительный синдром, индуцированный адъювантами (ASIA), Гемофагоцитарный лимфогистиоцитоз, болезнь Бехчета, Синдром Алажиля, Миопатия с тельцами включения с ранним началом болезни Педжета и лобно-височной деменцией (IBMPFD), Синдром Лоу, Синдром Питта Хопкинса, Синдром делеции 1p36, Метафизарная хондродисплазия типа Янсена, Синдром Коккейна, Хронический рецидивирующий многоочаговый остеомиелит, КРМО, Синдром Малана, Наследственная сенсорная и вегетативная нейропатия типа Ie, Заболевание ВКП, Гипнические подергивания, Миоклонус сна, Молларет Менингит, Рецидивирующий вирусный менингит, CRB1, Врожденный амавроз Лебера, Пигментный ретинит, Редкое заболевание сетчатки, KCNMA1-каннелопатия, Первичный билиарный цирроз, ZMYND11, Транзиторная глобальная амнезия, Болезнь накопления гликогена, Синдром Альстрема, Синдром Уайта Саттона, ДНМ1, EIEE31, Синдром Мире, Рецидивирующий респираторный папилломатоз, Папилломатоз гортани, Папилломатоз трахеи, Болезнь Рефсума, Синдром Николаидеса Барайтсера, лейкодистрофия, Танго2, Синдром конского хвоста, Редкие желудочно-кишечные расстройства, Ахалазия-Аддисоновский синдром, Ахалазия кардии, Синдром ахалазии икроцефалии, Анальный свищ, Врожденный дефицит сахаразы-изомальтазы, Эозинофильный гастроэнтерит, Идиопатический гастропарез, Болезнь Гиршпрунга, Редкое воспалительное заболевание кишечника, Кишечная псевдонепроходимость, склеродермия, Синдром короткой кишки, Сакральная агенезия, Синдром крестцовой агенезии, Каудальная регрессия, Болезнь Шейермана, Усеченные мутации SMC1A (вызывающие потерю функции гена), Цистиноз, Ювенильный нефропатический цистиноз, Нефропатический цистиноз, Кеннеди Болезнь, Спинно-бульбарная мышечная атрофия, Микросиндром Варбурга, Муколипидозы, Митохондриальные заболевания, Митохондриальные аминоацил-тРНК-синтетазы, Нарушения Mt-aaRS, Гипертрофическая оливарная дегенерация, Некетотическая гиперглицинемия, Синдром рыбного запаха, неприятный запах изо рта
Читайте также: