Недержание пигмента у новорожденного
Добавил пользователь Morpheus Обновлено: 27.01.2026
Синдром Блоха-Сульцбергера, или семейная форма недержания пигмента (Bloch-Sulzberger syndrome, familial incontinentia pigmenti)
Другие названия заболевания: синдром Асбо-Хансена, пигментный дерматоз Блоха-Сименса, нейрокожный меланобластоз.
Редкое наследственное заболевание кожи, подробно описанное швейцарским дерматологом Блохом в 1926 г. и американским дерматологом Сульцбергером в 1928 г. Болезнь наследуется по Х-сцепленному доминантному типу, т.е. характерна для представительниц женского пола, и обычно летальна для плодов мужского пола на ранних стадиях эмбриогенеза. Заболевание передается от матери или в результате возникновения мутации de novo (у 40% пациенток), как правило, в отцовских половых клетках. Описаны редкие случаи рождения мальчиков с синдромом Блоха-Сульцбергера, выживание которых объяснялось наличием в кариотипе дисомии по хромосоме Х (имели также синдром Кляйнфельтера) или соматическим мозаицизмом по мутантной хромосоме Х или 46,XY/47,XXY.
Главным признаком болезни является дерматоз, который обнаруживается уже при рождении или в первые дни и недели жизни ребенка, классически наблюдается в виде линий, пятен, брызг вдоль эмбриональных линий Блашко и развивается в 4 стадии: везикулобуллезная (воспалительная, пик начала – до 2-х недель жизни), гипертрофическая (2-6 недель), пигментная (пик начала – 12-26 недель, сохраняется до 5-20 лет) и атретическая. Помимо дерматоза 80% пациенток имеют различные аномалии развития зубов, 40% – патологию волос и ногтей, 30% – заболевания глаз неврологического характера и другие проявления расстройства центральной нервной системы. Степень проявления признаков синдрома очень варьирует среди больных, даже у членов одной семьи, поэтому строгих диагностических критериев для синдрома нет. Клинический диагноз может быть поставлен при наличии главного признака. Дополнительные признаки свидетельствуют в пользу диагноза, в том числе семейная история, не исключающая Х-сцепленное наследование заболевания, или история привычного невынашивания беременности.
Точный диагноз позволяет поставить молекулярно-генетическое исследование на наличие мутаций в гене IKBKG (или NEMO). Данный ген кодирует ключевую регуляторную субъединицу NEMO ингибиторной киназы IKK2 сигнального пути ядерного транскрипционного фактора NF-кВ, который контролирует иммунный ответ, ответ на стресс, воспалительную реакцию, клеточную адгезию и защиту от апоптоза. Наличие дефекта в субъединице NEMO приводит к отсутствию активации сигнального пути NF-кВ на все известные стимулы, чем объясняется повреждение тканей, экспрессирующих мутантный ген IKBKG у больных девочек с недержанием пигмента, а также ранняя смерть плода мужского пола с мутацией в гене IKBKG из-за процесса апоптоза в печени, индуцированного ФНО. Мутации в гене IKBKG обнаруживаются у 70-80% пациенток с недержанием пигмента. Известна одна повторяющаяся протяженная делеция экзонов 4-10, которая составляет 80-90% всех мутаций в гене IKBKG. Остальные мутации включают как точковые дефекты, так и другие протяженные перестройки гена. Степень проявления внешних признаков синдрома, в частности обширность и рисунок поражения кожи, зависит от индивидуального паттерна инактивации хромосом Х в тканях, подверженных изменениям.
Описанные выше мутации приводят к полной инактивации NF-кВ. Известны редкие мягкие точковые мутации в гене IKBKG, которые не полностью инактивируют NF-кВ, что позволяет выживать эмбрионам мужского пола, несущим мягкую мутацию. Фенотипически у таких мальчиков наблюдается не синдром Блоха-Сульцбергера, а гипогидротическая или ангидротическая эктодермальная дисплазия с иммунодефицитом (XL-HED/EDA-ID) либо с иммунодефицитом с остеопетрозом и лимфедемой (XL-OL-EDA-ID). Женщины с подобными мягкими мутациями в гене IKBKG либо ассиптоматичны, либо имеют слабо выраженные признаки недержания пигмента.
Дифференциальный диагноз: пиодермия, герпетиформный дерматит, токсидермия, крапивница, мастоцитоз, пигментный и веррукозный невусы, врожденный буллезный эпидермолиз, болезнь Дарье, точечная хондродисплазия Конради-Хюнермана, гипомеланоз Ито, витилиго, синдром Негели.
В Центре Молекулярной Генетики для молекулярной диагностики синдрома Блоха-
Сульцбергера у лиц женского пола проводится поиск наиболее частой протяженной делеции экзонов 4-10 гена IKBKG (или NEMO) у пробанда, родителей и членов семьи с признаками заболевания, также исследуется инактивация (лайонизация) хромосом Х. Для диагностики синдрома у мальчиков возможен поиск наиболее частой мутации в биоптате пораженной ткани!, а также исследование кариотипа, в том числе FISH.
Недержание пигмента. Описание клинического случая.
Недержание пигмента представляет несомненный интерес, так как является редким генетическим заболеванием, и знание его клинических особенностей необходимо для дифференциальной диагностики с другими дерматозами. Для данной патологии характерна комбинация кожных и внекожных аномалий. Приводится описание клинического случая синдрома Блоха-Сульцбергера у ребёнка в возрасте 1 месяца.
Ключевые слова
Статья
Недержание пигмента. Описание клинического случая.
Научный руководитель: профессор, д.м.н. Слесаренко Н.А.
ФГБОУ ВО Саратовский ГМУ им. В. И. Разумовского Минздрава России
Кафедра дерматовенерологии и косметологии
Цель работы: описать клинический случай редкой патологии «Недержание пигмента».
Среди наследственных заболеваний в детском возрасте значимое место занимают состояния, сопровождающиеся поражением кожи, на которые можно сразу обратить внимание даже у новорождённых. С этих позиций особый интерес вызывает заболевание «Недержание пигмента».
Недержание пигмента, или синдром Блоха-Сульцбергера в настоящее время представляет собой редкое генетическое заболевание, характеризующееся дисплазией наружного и среднего зародышевого листка, а также неспособностью базальных клеток эпидермиса удерживать меланин, в результате чего пигмент скапливается в меланофорах дермы и в межклеточном пространстве.
Первое описание данного синдрома было сделано английским врачом Garrod A. в 1906 г., а швейцарский дерматолог Bloch B. и американский педиатр Sulzberger M. более подробно описали и проанализировали клинические наблюдения (1926-27гг.).
Популяционная частота встречаемости заболевания 1 : 90000 новорождённых, среди которых более 90% детей являются лицами женского пола.
Данное заболевание является мозаичным, так как в организме присутствуют генетически различимые ткани из одной зиготы. Выявлены два локуса для недержания пигмента: Хq11 с аутосомной транслокацией и с Х-сцепленными доминантными мутациями в гене NEMO.
Для данного дерматоза характерна комбинация кожных и внекожных аномалий. Для кожных проявлений характерна стадийность течения:
I стадия – воспалительная (везикуло-буллезная), возникает при рождении или в первые дни жизни ребёнка и длится до 2 месяцев. На эритематозно-отечном кожном покрове появляются пузыри и пузырьки с плотной покрышкой и прозрачным содержимым, локализирующиеся линейно, чаще на сгибательных поверхностях конечностей, нередко на туловище, на лице и коже волосистой части головы, образующие очаги по линиям Блашко.Увеличение количества элементов сыпи растёт по мере возникновения повторных вспышек, которые длятся от нескольких дней до 3-5 недель. На месте вскрывшихся элементов образуются эрозии с серозными корками. Высыпания появляются приступообразно. Слизистые оболочки не поражаются. Общее состояние ребенка не страдает. На этой стадии заболевания может обнаруживаться выраженная эозинофилия (до 40-50%) в периферической крови.
II стадия – гипертрофическая (папуловеррукозная), развивается спустя 2-3 месяца. Проявляется в виде чечевицеобразных ороговевающих папул, которые локализуются линейно и дугообразно (по линиям Блашко) в области бывших элементов первой стадии, или беспорядочно, имея схожее строение с бородавчатым невусом желто-бурого цвета. На ладонях и подошвах возможен диффузный гиперкератоз. Подобные изменения кожи сохраняются в течение нескольких месяцев.
III стадия – пигментная, возникает в возрасте 5–6 мес. Бородавчатые очаги разрешаются с образованием гиперпигментированных пятен серо-коричневого цвета со светлыми краями. Рисунок кожных аномалий может иметь самые необыкновенные очертания: на конечностях высыпания похожи на «брызги грязи», на туловище – на «спирали», «кольца» или «мраморный кекс». Чаще всего пигментация начинает исчезать в возрасте 5-6 лет, но может сохраняться и у взрослых (до 10-30 лет).
IV стадия – атрофическая, развивается не у всех пациентов и проявляется через 20-30 лет жизни. Характеризуется сменой участков пигментации на депигментации, умеренно выраженной атрофией кожи, очаговым склерозом.
В настоящее время выделяют 2 формы синдрома Блоха-Сульцбергера:
I форма - стадийная (описана выше);
II форма - воспалительная стадия отсутствует: у ребёнка в 2-3-х летнем возрасте появляются бородавчатые разрастания по линиям Блашко, часто принимаемые за невусы. Они существуют долго, до пубертатного периода и даже дольше.
Внекожные изменения встречаются почти у 50% больных и чаще всего проявляются аномалиями со стороны зубов (65%), органов зрения (35%), ЦНС (16%), а также сердечно-сосудистой системы, опорно-двигательного аппарата и мочевыделительной системы.
Дифференциальная диагностика зависит от стадии заболевания. При I стадии исключают буллёзные дерматозы: простой пузырьковый лишай новорождённых, стафилококковую (эпидемическую) и сифилитическую пузырчатки, буллезный эпидермолиз. При II и III стадиях дифференцируют с невусами, дискератозом Дарье, вульгарными бородавками, доброкачественной пузырчаткой Хейли-Хейли.
Прогноз при «Недержании пигмента» зависит от стадии и вовлечения в процесс различных органов и систем.
Лечение больных симптоматическое. В период воспалительных явлений для профилактики присоединения инфекции используется наружная терапия водными анилиновыми красителями, антисептическими растворами и присыпками. При генерализации процесса возможно назначение дезинтоксикационных средств, системных антибиотиков, глюкокортикостероидов. В стадии веррукозных разрастаний рекомендованы кератолитические препараты, наружные или системные ароматические ретиноиды.
Клинический случай. Больная М. в возрасте 1 месяца поступила в детскую инфекционную больницу с диагнозом «Инфекция неясной этиологии». Из анамнеза известно, что ребёнок от 2-ой беременности, первая беременность закончилась выкидышем плода мужского пола. В I триместре мать отмечала у себя на бедре сыпь наподобие герпетической, лечение не проводилось. Роды срочные, естественным путём. Масса ребёнка при рождении 3400 г., рост - 52 см, состояние расценивалось как удовлетворительное. Через несколько часов кожа приобрела ярко розовый цвет, на верхних и нижних конечностях появились сгруппированные пузырьки и пузыри диаметром от 0,5 до 2 см с серозным содержимым. По данному поводу была переведена в отделении патологии новорожденных с диагнозом «Внутриутробная инфекция?». Далее с диагнозом «Инфекция неясной этиологии» была направлена в детскую инфекционную больницу для уточнения диагноза и лечения.
Объективно: поражение кожи имеет распространенный характер. На коже нижних и верхних конечностей на ярко-эритематозном фоне отмечаются множественные линейно расположенные, по линиям Блашко, везикуло-буллёзные высыпания размером от 0,3 до 1,5 см с гладкой напряженной покрышкой, с серозным содержимым. Уплотнения в основании пузырей не определяются. На коже бедра правой нижней конечности и подколенной ямки левой нижней конечности высыпания образуют бляшки, покрытые серозно-геморрагическими корками.
Инфекционный характер заболевания был отвергнут в первые дни пребывания в инфекционной больнице. Дифференциальный диагноз проводился с буллёзным эпидермолизом и линеарным IgA- зависимым дерматозом у детей.
Ребёнок получал антибактериальную терапию с целью профилактики присоединения бактериальной инфекции. Наружно назначались антисептические растворы и анилиновые красители.
На фоне проводимого лечения отмечено значительное улучшение: эритема угасла, экссудативные элементы подсохли, корки отторглись.
Было рекомендовано продолжить обследование: консультация невролога, офтальмолога, кардиолога, дерматолога и педиатра.
Вывод: таким образом, новорожденные с везикуло-буллёзными высыпаниями, наблюдаемые неонатологами, требуют тщательного обследования и определения дальнейшей тактики ведения.
Литература
1. Воинова В.М. Российский вестник перинатологии и педиатрии. Синдром Блоха-Сульцбергера у детей/ В.М. Воинова, П.В. Новиков, Л.З. Казанцева. – Москва, 1999.- 25-28с.
2. Слесаренко Н.А. Саратовский научно-медицинский журнал. Недержание пигмента (синдром Блоха - Сульцбергера)/ Слесаренко Н.А. [и д.р.] – Саратов, 2015.- 457–462с.
3. Прошутинская Д.В. Вестник дерматологии и венерологии. Невоидная гипопигментация как проявление мозаицизма кожи/ Д.В. Прошутинская, Л.В. Текучева. - Москва, 2015.- 86-90с.
4. Вислобоков А.В. Российский журнал кожных и венерических болезней. Меланобластоз Блоха–Сульцбергера/ Вислобоков А.В. [и д.р.] – Москва, 2015.- 26-29с.
Недержание пигмента у новорожденного
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
Недержание пигмента (синдром Блоха−Сульцбергера)
Журнал: Клиническая дерматология и венерология. 2016;15(3): 61‑63
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
Описан системный генодерматоз (синдром Блоха—Сульцбергера), характеризующийся своеобразными стадийными изменениями кожи, сочетающимися с поражением волос, ногтей, зубов, центральной нервной и костно-мышечной систем. Приведено собственное клиническое наблюдение за пациенткой в возрасте 4 мес.
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
ГБОУ ВПО «Волгоградский государственный медицинский университет», Волгоград, Россия, 400120
Недержание пигмента (синдром Блоха—Сульцбергера) — системный меланобластоз, характеризующийся специфическими стадийными изменениями кожи в сочетании с поражением центральной нервной системы, волос, ногтей, глаз, зубов, костно-мышечного аппарата, встречающийся с частотой 1:75 000 [1—3]. Пациент с данной патологией впервые был продемонстрирован в 1906 г. A. Garrod. Позже В. Bloch (1925) и его ученик М. Sulzberger (1928), описав клинические проявления, дали название болезни — недержание пигмента [2, 3]. Причинами недержания пигмента считают травмы и вирусные инфекции матери во время беременности, приводящие к внутриутробной аллергизации и интоксикации плода, нестабильность хромосом, аутоиммунные нарушения, приводящие к повреждению клеток эктодермального происхождения, имеющих аномальные поверхностные антигены [1, 2, 4]. Заболевание обусловлено мутацией гена NEMO, картированного на участке Xq28, имеет Х-сцепленное доминантное наследование с внутриутробной гибелью плодов мужского пола, поэтому болеют практически исключительно девочки [1, 5]. Выявление патологии у мальчиков расценивают как развитие спонтанной мутации [6].
Клиническая картина в своем развитии проходит четыре стадии.
— I стадия (воспалительная или везикуло-буллезная) характеризуется появлением пузырьков, пузырей, эритематозных уртикароподобных распространенных высыпаний сразу после рождения или в первые 2 нед жизни ребенка. Высыпания разрешаются и одновременно появляются вновь. Из лабораторных показателей обращает на себя внимание эозинофилия в общем анализе крови и содержимом пузырей [1]. Диагностика синдрома недержания пигмента в данной стадии порой бывает достаточно трудной [7].
— II стадия (веррукозная) развивается обычно в возрасте 2—3 мес, иногда даже без предшествующих изменений кожи, и характеризуется появлением узелков с гиперкератотическими веррукозными разрастаниями. На ладонях и подошвах может быть выраженный гиперкератоз [1].
— III стадия (гиперпигментация) обычно диагностируется в 5—6 мес. Для нее характерно появление пигментных пятен с сетчатым ветвистым рисунком коричневато-желтого или сероватого цвета с четкими границами, которые обычно сравнивают с «брызгами грязи», «завихрениями», «звездчатым узором» [1, 2].
— IV стадия (гипопигментация и атрофия) — очаги гиперпигментации сменяются очагами гипопигментации, иногда с поверхностной атрофией.
Проявления, характерные для различных стадий, могут сочетаться одновременно у одного больного. Помимо кожных проявлений, у пациентов с недержанием пигмента могут быть нарушения нервной системы в виде задержки психомоторного развития, эпилептиформных припадков, гидроцефалии; патология глаз вплоть до атрофии зрительного нерва и слепоты, аномалии скелета, дистрофии зубов, волос, ногтей [1, 8—10].
Ранее нами был описан случай синдрома Блоха—Сульцбергера в стадии гиперпигментации у девочки 7 мес, при этом у матери и бабушки ребенка обнаруживали гипопигментированные пятна в области нижних конечностей [11].
Для демонстрации трудностей диагностики и возможных диагностических ошибок у детей грудного возраста приводим собственное наблюдение.
Больная В., 4 мес, родилась 24.02.12 в срок с массой тела 2750 г. От седьмой беременности четвертым ребенком в семье (есть старшая сестра 11 лет и два брата 8 и 5 лет). Мальчики здоровы. У старшей сестры наблюдались симптомы нарушения пигментации в раннем детском возрасте, которые довольно быстро разрешились, и в настоящее время девочка практически здорова. У матери проявлений заболевания не выявлено. У младшей девочки сразу после рождения были эритематозные высыпания на коже, которые довольно быстро регрессировали после курса антибактериальной терапии. После выписки из родильного дома стали появляться буллезные высыпания на фоне эритемы. В возрасте 1 мес присоединились веррукозные гиперкератотические высыпания. Дерматологом по месту жительства был выставлен диагноз «веррукозный невус». Девочка отставала в психомоторном развитии. В возрасте 4 мес после консультации генетика выставлен диагноз «Наследственный синдром, сопровождающийся нарушением нервно-психического развития». При осмотре в клинике: в области голеней, тыла стоп, кистей имеются линейно расположенные папулы, веррукозные очаги розового и красного цветов с выраженными гиперкератотическими наслоениями, сетчатые и линейные очаги гиперпигментации, на подошвах и ладонях очаги гиперкератоза (рис. 1, 2, 3). Слизистые оболочки, волосы, ногти не изменены, аномалий скелета не выявлено, отмечается задержка психомоторного развития. Ребенку был выставлен диагноз «Недержание пигмента, веррукозная стадия».
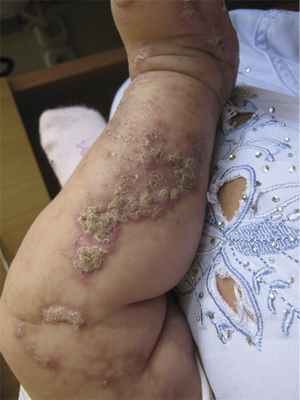
Рис. 1. Веррукозные очаги и участки сетчатой гиперпигментации в области голеней.
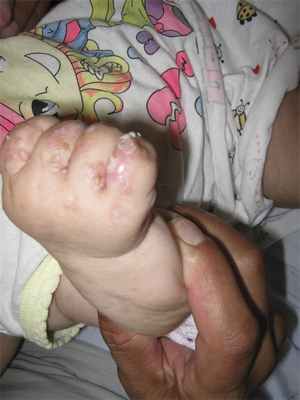
Рис. 2. Папулы с веррукозными разрастаниями на поверхности.
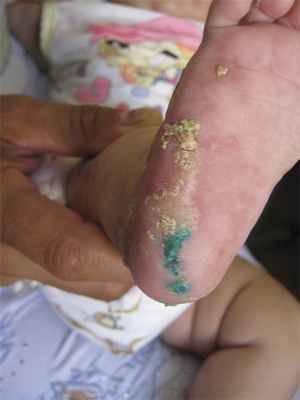
Рис. 3. Очаги гиперкератоза в области подошв.
Критериями диагностики недержания пигмента в данном случае являлись: анамнестические данные (развитие высыпаний у обеих сестер сразу после рождения), появление вначале буллезных, а затем гиперкератотических высыпаний на коже, очагов гиперпигментации, задержка психомоторного развития. В данном случае, по нашему мнению, речь может идти об особом варианте недержания пигмента — синдроме Асбо−Хансена (буллезный кератогенный пигментный дерматит) [2]. Для этого типа характерны папуло-веррукозные и буллезные высыпания, расположенные линейно (обычно на конечностях), на фоне которых в дальнейшем появляются очаги гиперпигментации.
Интерес данного клинического случая обусловлен четким развитием заболевания в семье только у девочек, наличием у ребенка веррукозных высыпаний наряду с имеющимися очагами гиперпигментации, что подтверждает данные некоторых авторов о возможном сочетании признаков разных стадий синдрома Блоха—Сульцбергера [1, 7].
Тяжелая форма синдрома Блоха—Сульцбергера у новорожденного ребенка
Диагностика заболеваний кожи у новорожденных нередко является сложной междисциплинарной проблемой. В статье приведены данные литературы и собственное клиническое наблюдение новорожденного ребенка с редким генным дерматозом — синдромом Блоха—Сульцбергера. Особенностью наблюдения явилось развитие заболевания сразу после рождения ребенка, сложная дифференциальная диагностика. Вовлечение в процесс центральной нервной системы в виде эпилептического синдрома обусловливает тяжесть состояния пациента и серьезно влияет на прогноз течения заболевания. Обсуждаются вопросы углубленных исследований с использованием молекулярно-генетических технологий, повышающих ценность медико-генетического консультирования семьи.
Ключевые слова
Об авторах
Детская областная клиническая больница им. З.И. Круглой, Орел
Россия
врач-неона-тологинфекционно-боксированного отделения для новорожденных Детской областной клинической больницы им. З.И. Круглой
зав. инфекционно-боксированным отделением для новорожденных той же больницы
врач-генетик той же больницы; асе. каф. педиатрии с курсом детской хирургии Орловского медицинского института
д.м.н., проф. той же каф. 302019 Орел, ул. Октябрьская, д. 4
Список литературы
1. Хегер Петер Г. Детская дерматология. Пер. с нем. Под ред. А.А. Кубановой, А.Н. Львова. М: БИНОМ. Лаборатория знаний 2013; 77—78. (Heger P.G. Pediatric dermatology. Trans, from Ger. A.A. Cubanova, A.N. Lvov (eds.). M: BINOM. Knowledge Laboratory 2013; 77—78.)
2. Воинова В.М., Новиков П.В., Казанцева Л.Н. Синдром Блоха—Сульцбергера у детей. Рос вестн перинатол и педиат 1999; 5: 25—28. (Voinova V.M., Novikov P.V., Kazantseva L.N. Bloch-Sulzberger syndrome in children. Ros vestnperinatal i pediat 1999, 5: 25—28.)
3. Детская дерматология. Справочник под ред. Д.П. Кроу-гука, А.Дж. Мангини, пер. с англ. Под ред. H.L. Короткого. М: Практ мед 2010; 468—473. Pediatric Dermatology.
4. A Handbook. Transl. from English. N.G. Korotkij (ed.). M: Prakti med 2010; 468—473.)
5. Зверькова Ф.А. Болезни кожи детей раннего возраста. Ст-Петербург 1994; 131—135. (Zverkova F.A. Diseases of the skin in infants. St-Petersburg 1994; 131—135.)
7. Козлова СИ., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М: Т-во научных изданий КМК; Авторская академия, 2007; 179—181. (Kozlova S.I.,DemikovaN.S. Hereditary syndromes andgenetic counseling. M: T-vo nauchnykh izdanij KMK; Avtorskaya akademiya, 2007; 179-181.)
Рецензия
Для цитирования:
Черникова Т.И., Шепилов Л.А., Васина Т.Н., Зубцова Т.И., Ставцева С.Н., Вислобоков А.В. Тяжелая форма синдрома Блоха—Сульцбергера у новорожденного ребенка. Российский вестник перинатологии и педиатрии. 2014;59(5):59-62.
For citation:
Chernikova T.I., Shepilov L.A., Vasina T.N., Zubtsova T.I., Stavtseva S.N., Vislobokov A.V. Severe Bloch—Sulzberger syndrome in a newborn baby. Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics). 2014;59(5):59-62. (In Russ.)
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Недержание пигмента у новорожденного
Ф. Каммарата-Скалиси, Отдел медицинской генетики, педиатрии, университет Анд, Мерида, Венесуэла; Ф. Фуско, М.В. Урсини, Институт генетики и биофизики «Адриано Буццати-Траверсо», IGB-CNR, Неаполь, Италия
Введение
Недержание пигмента (OMIM 308300), редкая нейроэктодермальная дисплазия (1, 2), является Х-сцепленным доминантным заболеванием, вызванным мутациями в гене IKBKG/NEMO (GenBankNM_003639.3, OMIM 300248), ранее известном как NEMO.
Ген расположен на Xq28 и кодирует легкий полипептидный энхансер гена kappa в В-клетках, киназу гамма, которая играет ключевую роль в модуляции ядерного фактора транскрипции kappa B (NF-kB) (1, 3-7).
Недержание пигмента было описано Блохом в 1926 году и Сульцбергером в 1928 (4) году и поэтому также известно, как синдром Блоха–Сульцбергера (6, 8).
Ответственный за развитие заболевания ген обладает высокой пенетрантностью и переменной экспрессивностью (4).
Делеция 11,7 к b , охватывающая экзоны от 4 до 10, является основной причиной недержания пигмента и встречается примерно в 80% случаев (3, 6, 9, 10).
Это вызвано рекомбинацией между двумя повторами MER67B в интронах 3 и 10 гена, что приводит к потере функции белка. Также были описаны небольшие инсерционно-делеционные и нонсенс-мутации.
Некоторые гипоморфные мутации IKBKG/NEMO вызывают отчетливые изменения: гипогидротическую эктодермальную дисплазию с иммунодефицитом (OMIM 300291) (6, 11).
Эпидемиология
По оценкам, частота встречаемости недержания пигмента составляет 0,7 случая на 100 000 родов (10) и 27,6 новых случаев ежегодно. От 65% до 75% случаев обусловлены спорадическими мутациями, а остальные случаи являются семейными (2).
Это состояние обычно приводит к летальному исходу у гемизиготных мужских эмбрионов (2-4, 12-13).
Пациенты женского пола могут выжить благодаря функциональному мозаицизму, возникающему в результате инактивации Х-хромосомы (2, 4, 9). Клинические проявления в таких случаях весьма разнообразны (3).
Таким образом, недержание пигмента преобладает у женщин, причем соотношение женщин и мужчин составляет 37:1. Пациенты мужского пола с клиническими признаками недержания пгмента также могут иметь синдром Клайнфельтера (47, XXY).
Выживаемость в таких случаях возможна за счет второй хромосомы Х или соматического мозаицизма для упомянутой выше общей делеции (4, 5, 8).
Клинические особенности и ведение пациентов
Недержание пигмента – это мультисистемное заболевание (4, 5, 8), поражающее как эктодермальные, так и мезодермальные ткани. Кожа всегда вовлечена и её поражение является основным диагностическим критерием (9).
Дополнительные изменения затрагивают центральную нервную систему, глаза, зубы (3, 4, 10, 12), молочные железы, волосы, ногти и скелет. Сердечно-легочные изменения также наблюдаются, хотя и реже (12).
Лэнди и Доннаи (14) установили первые диагностические критерии недержания пигмента в 1993 году, еще до открытия ответственного гена (10).
В 2014 году Minić и соавт. (15) предложили ряд обновленных критериев, а в 2018 году, с разрешения авторов, Россер (3) модифицировал эти критерии для улучшения фенотипической характеристики заболевания и дальнейшего обновления диагностических критериев.
Основными предложенными критериями были типичные дерматологические проявления, наблюдаемые при недержании пигмента, которые используются для установления клинического диагноза (3, 5), в то время как второстепенные критерии соответствовали другим возможным проявлениям.
Дополнительные диагностические критерии включают положительный семейный анамнез (у родственника первой степени родства) и мутацию в гене IKBKG/NEMO.
Они описаны в таблице 1 (2-20) вместе с другими критериями, описанными Россером (3), и соответствующими частотами встречаемости, описанными в литературе.
Таблица 1.Предложенные диагностические критерии недержания пигмента и распространенность некоторых соответствующих состояний, о которых сообщается в литературе
Поражения кожи являются наиболее ярким проявлением. Они появляются при рождении или в первые 2 недели жизни, прогрессируют в течение нескольких лет и распределяются по линиям Блашко.
Существует 4 классические стадии: везикобуллезная стадия (рис. 1), веррукозная/гиперкератотическая стадия, гиперпигментированная стадия (рис. 2) и атрофическая/гипопигментированная стадия (19, 20). Не все пациенты имеют 4 стадии (19), первые 3 могут появиться одновременно (20).
Эти поражения более подробно описаны в таблице 1 (2-20). Термин «недержание пигмента» связан с гистологическими характеристиками поражений на 3-й стадии заболевания (гиперпигментация) и, в частности, с недержанием меланина меланоцитов в базальном слое эпидермиса и отложений в поверхностной дерме (4).
Эти проявления, как правило, не требуют специального лечения (3). Лечение топическими кортикостероидами или такролимусом может замедлить прогрессирование пузырно-буллезной стадии, хотя поражения регрессируют спонтанно (18).
Недержание пигмента в значительной степени становится серьезным при наличии неврологических изменений (9, 10), так как это может привести к смерти (19).
Неврологические нарушения разнообразны по своей природе, но наиболее частыми являются припадки (9, 21), которые, как правило, тонические (22) или фокально-клонические, длятся недолго и вызывают потерю сознания.
Они чаще встречаются в неонатальном периоде (9), устойчивы к лечению и имеют плохой прогноз.
Другие нарушения включают микроцефалию, ишемический инсульт, мозжечковую атаксию и задержку психомоторного развития. Острые проявления энцефалита в основном появляются в первые 4 дня жизни и включают пищевую непереносимость, вялость и судорожные движения.
Таким образом, магнитно-резонансная томография головного мозга с ангиографией имеет важное значение у новорожденных с характерными кожными проявлениями недержания пигмента. Последующие исследования также необходимы для предотвращения последствий (3, 9, 22).
Нарушение зрения наблюдается у 50-77% пациентов. Наиболее серьезные изменения затрагивают сетчатку, и такие заболевания сетчатки (16), как отслойка и аваскулярная сетчатка, являются наиболее опасными осложнениями.
Их можно лечить с помощью лазерной фотокоагуляции, направленной на неоваскуляризацию сетчатки. Криотерапия может быть также рекомендована; хорошие результаты наблюдаются при применении ингибиторов сосудистого эндотелиального фактора роста (18).
Кадры оптической когерентной томографии показывают истончение внутреннего и внешнего слоев сетчатки. Аномалии глаз, сходные с ретинобластомой, были описаны в некоторых случаях недержания и включают лейкокорию, косоглазие, внутриглазную кальцификацию и, как уже упоминалось, отслойку сетчатки.
Поэтому очень важен дифференциальный диагноз (16). Пациенты должны проходить обследование глаз каждый месяц в течение первые 4 месяца жизни, каждые 3 месяца до 1 года, каждые 6 месяцев до 3 лет и каждые 12 месяцев последующие периоды (18).
Этиология и патогенез
NF-kB активируется белковым продуктом IKBKG/NEMO, который играет ключевую модулирующую роль в различных физиологических функциях, таких как иммунный ответ и стресс, воспалительные реакции, эктодермальное развитие, клеточная адгезия и защита клеток от апоптоза, индуцированного фактором некроза опухоли (3, 4, 6, 22).
Мутация IKBKG/NEMO приводит к снижению активности NF-kB, тем самым повышая восприимчивость к апоптозу (4). Обширный апоптоз у мужчин ответственен за раннюю гибель плода (10) и нарушения функции печени (2).
Воспалительные реакции и эпидермальный рекрутинг эозинофилов, возникающие на первой стадии заболевания, как правило, играют важную патогенетическую роль (4).
Это, вероятно, связано с эозинофильным селективным хемокином (эотаксином) (4, 16), который продуцируется специфическими лейкоцитами, такими как эозинофилы, макрофаги и Т-клетки, а также структурными клетками, такими как эндотелиальные клетки, фибробласты и эпителиальные клетки (4).
Дифференциальная диагностика
Признаки, которые следует учитывать при дифференциальной диагностике, различаются в зависимости от стадии заболевания. На первой стадии недержание пигмента можно спутать с инфекциями новорожденного, такими как врожденный простой герпес, ветряная оспа, буллезный эпидермолиз и буллезный пемфигоид (18, 23).
Поскольку неонатальный герпес и недержание пигмента могут сосуществовать, лечение ацикловиром необходимо начинать сразу же после подтверждения вирусной инфекции. Дифференциальная диагностика на втором этапе ограничена и должна включать линейный эпидермальный невус (23).
Третья стадия является характерной стадией недержания пигмента и должна быть дифференцирована от линейного и невоидного гипермеланоза. Четвертая стадия, если она присутствует, может быть ошибочно принята за гипомеланоз Ито или витилиго. Эти состояния, однако, не имеют начальной воспалительной фазы и, как правило, не влияют на придатки кожи (18).
Выводы и генетическое консультирование
Диагноз недержания пигмента основывается на клинических данных (18).
Тем не менее, учитывая огромную вариабельность клинических признаков и широкий спектр молекулярных изменений, трудно выделить однородную группу пациентов и стандартизировать лечение.
Несмотря на прогресс, достигнутый в нашем понимании причин недержания пигмента и в клинических исследованиях, нехватка пациентов, наблюдаемых в диагностических центрах, затрудняет получение глобальной эпидемиологической картины.
Интеграция этих данных может внести важный вклад в будущие научные усилия (10).
Когда речь идет о мультисистемном заболевании, необходимо длительное, индивидуальное, мультидисциплинарное наблюдение с оценкой состояния дерматологами, неврологами, офтальмологами и стоматологами, среди прочих (3, 24).
Врожденный гипотиреоз, миастения и опухоль Вильмса были описаны в некоторых случаях недержания пигмента, и одна группа авторов, которая недавно наблюдала заболевание печени у пациента с недержанием пигмента, предложила включить оценку печени в эти условия, хотя они не смогли установить связь между этими двумя заболеваниями (1).
Это может означать, что пациентам с недержанием пигмента может потребоваться еще более широкое обследование.
Кроме того, обновленные диагностические критерии, предложенные Россером, не включают в себя скелетные изменения (например, низкий рост, гемивертебра, кифоз, сколиоз, лишние ребра, дисплазия тазобедренного сустава, гемиатрофия, косолапость (4) или синдактилия пальцев ног (4, 10)) или сердечно-легочные нарушения (например, дефект межпредсердной перегородки (4), эндомиокардиальный фиброз левого желудочка, трикуспидальная недостаточность, тетрада Фалло (4, 17) или легочная гипертензия, даже при отсутствии сердечно-сосудистых изменений) (2, 4).
Риск рецидива инфекции также в настоящее время не рассматривается (10). Будущие обновления, вероятно, будут включать многие из этих заболеваний, чтобы охватить основные клинические проявления этого заболевания.
Генетические исследования необходимы для углубления нашего понимания недержания пигмента и подтверждения диагноза с помощью молекулярных методов диагностики, особенно в сомнительных случаях (18).
Прогноз в целом хороший, но, как уже упоминалось, важно регулярное иеждисциплинарное наблюдение. Генетическое семейное консультирование также важно для пациентов с Х-сцепленным доминантным заболеванием (18, 23), которое обычно приводит к летальному исходу у пациентов мужского пола.
Пациенты женского пола в этом случае имеют 50%-ный риск родить дочь с недержанием пигмента. Выкидыш происходит примерно в 50% случаев плодов мужского пола. Выжившие мальчики должны быть направлены на цитогенетическое исследование.
Читайте также: