Обзор кожных порфирий
Добавил пользователь Владимир З. Обновлено: 09.01.2026
Порфирии - это редкие нарушения при которых аномалии метаболизма гемоглобина обусловлены генетической или приобретенной недостаточностью ферментов биосинтеза гема, что приводит к накоплению его токсичных предшественников. Эти недостатки позволяют предшественникам гема накапливаться, вызывая токсические реакции. В основе разных порфирий лежит недостаточность разных ферментов. Двумя основными проявлениями порфирий являются нейровисцеральные симптомы (при острых порфириях) и светочувствительность кожи (при кожных порфириях).
Гем, железосодержащий пигмент, является важным кофактором многих гемопротеинов. Практически всем клеткам человеческого тела требуется синтеза гема. Гем в основном синтезируется в костном мозге (эритробластами и ретикулоцитами) и включается в структуру гемоглобина. Печень является 2-м наиболее активным местом синтеза гема, который преимущественно включается в состав ферментов системы цитохрома Р-450. Для синтеза гема требуется 8 ферментов (см. таблицу Субстраты и ферменты биосинтеза гема [Substrates and Enzymes of the Heme Biosynthetic Pathway] Субстраты и ферменты биосинтеза гема и заболевания, связанные с их дефицитом ). Эти ферменты образуют и модифицируют молекулы, называемые порфириногенами или порфиринами (и их предшественниками). Накапливаясь, эти вещества вызывают клинические проявления порфирий.
Этиология порфирий
За исключением спорадического типа поздней кожной порфирии Поздняя кожная порфирия Поздняя кожная порфирия (ПКП) – сравнительно частая печеночная порфирия, при которой поражается преимущественно кожа. Также нередко имеют место заболевания печени. ПКП связана с приобретенной. Прочитайте дополнительные сведенияГомозиготные или комбинированные гетерозиготные (т.е. 2 отдельные гетерозиготные мутации, по одной в каждом аллели одного и того же гена, у одного больного) аутосомно доминантной порфирии могут быть несовместимы с жизнью, как правило, приводя к смерти плода. Гетерозиготные порфирии имеют различную степень пенетрантности, поэтому не все генетические случаи порфирии проявляются клинически. Из 2-х наиболее распространенных порфирий, острая перемежающаяся порфирия (ОПП), является аутосомно-доминантной и около 20% случаев ПКП также являются аутосомно-доминантными. Распространенность ПКП составляет около 1 случая на 10000 человек населения (1/10000). Распространенность генетической мутации, вызывающей ОПП, составляет около 1/1500, потому ее пенетрантность низкая, и распространенность клинических проявлений заболевания составляет около 1/10000. Распространенность ПКП и ОПП в различных регионах и этнических группах весьма различная.
При аутосомно-рецессивных порфириях заболевание вызывают только гомозиготные или комплексные гетерозиготные варианты. Третьей по распространенности порфирией является эритропоэтическая протопорфирия, относящаяся к аутосомно-рецессивному типу.
Одна из врожденных порфирий, Х-хромосомная протопорфирия, является результатом связывания с Х-хромосомой Эритропоэтическая протопорфирия и связанная с Х-хромосомой протопорфирия Эритропоэтическая протопорфирия (ЭПП) обусловлена наследственной недостаточностью фермента феррохелатазы. Х-сцепленная протопорфирия (X-ПП) связана с наследственным увеличением активности синтазы-2. Прочитайте дополнительные сведенияПатофизиология порфирий
Порфирии развиваются при недостаточности любого из последних 7 ферментов биосинтеза гема или из-за повышенной активности эритроидной формы первого фермента этого процесса – АЛК-синтазы-2 (АЛКС 2). (Дефицит ALAS 2 вызывает сидеробластную анемию Сидеробластные анемии Сидеробластные анемии представляют собой разнородную группу анемий, которые характеризуются увеличением сывороточного железа, ферритина и насыщения трансферрина также, как и наличием кольцевидных. Прочитайте дополнительные сведенияХотя порфирии наиболее точно определяются в соответствии с дефицитом фермента, часто бывает полезна классификация по основному месту гиперпродукции предшественников гема (гепатоциты или эритроциты) или основным клиническим признакам (острым или кожным).
Острые порфирии Острые порфирии Острые порфирии обусловлены дефицитом некоторых ферментов биосинтеза гема, приводящим к накоплению предшественников гема, которые вызывают периодические приступы боли в животе и неврологические. Прочитайте дополнительные сведения проявляются перемежающимися приступами с абдоминальной, психической и неврологической симптоматикой. В типичных случаях такие приступы провоцируются медикаментозными препаратами, циклическими изменениями активности гормонов у молодых женщин и другими экзогенными факторами. Кожные порфирии Обзор кожных порфирий Кожные порфирии обусловлены дефицитом (а в одном случае – избытком) некоторых ферментов пути биосинтеза гема (см. таблицу Субстраты и ферменты биосинтеза гема), приводящим к сравнительно постоянной. Прочитайте дополнительные сведения обычно сопровождаются длительными, периодически меняющимися симптомами, включая повышенную светочувствительность кожи. При некоторых острых порфириях (наследственной копропорфирии, смешанной порфирии) также возможны кожные проявления. В силу вариабельной пенетрантности гетерозиготных порфирий их клиническая частота меньше генетической (см. таблицу Основные свойства двух наиболее частых порфирий Основные свойства двух наиболее частых порфирий ).
В симптоматической фазе всех порфирий, за исключением эритропоэтической протопорфирии (ЭПП) и ДАЛК-дефицитной порфирии, моча приобретает красный или красно-коричневый цвет. Это обусловлено окислением порфириногенов и их предшественника порфобилиногена (ПБГ). Иногда патологическая окраска мочи развивается лишь после ее оставления на воздухе или свете в течение от нескольких минут до нескольких часов, что зависит от скорости неферментативного окисления. При острых порфириях, за исключением ALAD-дефицитной, примерно у 1 из 3 гетерозигот (у женщин чаще, чем у мужчин) экскреция ПБГ с мочой (с ее соответствующей окраской) повышена даже во время латентной фазы.
Диагностика порфирий
Исследование крови или мочи
При подозрении на порфирию определяют содержание предшественников порфиринов –порфобилиногена (ПБГ) и дельта-аминолевулиновой кислоты (АЛК) - в крови и моче (см. Скрининг на порфирии [Screening for Porphyrias] Скрининг на порфирии ). Аномальные результаты таких определений служат поводом для дальнейших исследований.
Подобным же образом обследуют и возможных носителей генетического дефекта, а также пациентов в период между приступами, т.е. в отсутствие симптомов заболевания. Однако в этих случаях содержание АЛК и ПБГ – гораздо менее чувствительный показатель, чем ферментативная активность эритроцитов и лейкоцитов. Тем не менее, тесты для определения многих ферментов пути (например, уропорфириноген-III-косинтазы [уроген-3-синтазы], копропорфириногеноксидазы [КПО], протопорфириногеноксидазы [ППО], феррохелатазы [ФХ]), как правило, не обще или коммерчески доступны.
Если мутация известна, то самые точные результаты при обследовании членов семьи больного дает генетический анализ. Генетическое тестирование выявит известные связанные с болезнью мутации у большинства пациентов с наследственными формами порфирии; тем не менее, у небольшого процента (~ 1%) пациентов с клиническими и биохимическими нарушениями генетическое тестирование не сможет выявить причинную мутацию. Таким образом, для постановки правильного диагноза все ещё требуется глубокая интеграция результатов клинических, биохимических и генетических исследований. Пренатальная диагностика (с помощью амниоцентеза или исследования ворсин хориона) возможна, но показана лишь в редких случаях.
Вторичная порфиринурия
Повышенной экскрецией с мочой порфиринов могут сопровождаться некоторые заболевания, не связанные с порфириями; это явление называют вторичной порфиринурией.
К повышенной экскреции копропорфирина с мочой могут привести гематологические заболевания, заболевания гепатобилиарной системы, токсины (например, алкоголь, бензол, свинец). Повышенная экскреция копропорфирина с мочой может развиваться при любых гепатобилиарных заболеваний потому, что один из путей экскреции порфирина - выделение с желчью. Большое количество лекарственных препаратов и химических веществ ингибирует переносчики органических анионов, которые обычно переносят порфирины, особенно копропорфирины, в желчь; к наиболее распространенным относятся артесунат, балсалазид, беназеприл, хлорпропамид, кортизол, демеклоциклин, дифлунизал, флавоноиды, ирбесартан, мефенаминовую кислоту, нитазоксанид, пенцикловир, пробенецид, стирипентол, телмисартан, валсартан и другие ( 1, 2 Справочные материалы касающиеся вторичной порфиринурии Порфирии - это редкие нарушения при которых аномалии метаболизма гемоглобина обусловлены генетической или приобретенной недостаточностью ферментов биосинтеза гема, что приводит к накоплению. Прочитайте дополнительные сведения ). Прием препаратов из этой группы также может увеличить экскрецию порфирина с мочой. При гепатобилиарных заболеваниях также может быть повышен уровень уропорфирина. Протопорфирин не выводится с мочой, поскольку нерастворим в воде.
Заболевания, которые вызывают вторичную порфиринурию (а также заболевания, которые вызывают клинические синдромы, имитирующие острые порфирии), как правило, не повышают уровни АЛК и ПБГ в моче, поэтому нормальные уровни АЛК и ПБГ помогают отличить вторичную порфиринурию от острых порфирий. Тем не менее, в некоторых случаях отравления свинцом Интоксикация свинцом При отравлении свинцом симптомы незначительны в начале, однако в дальнейшем могут развиться острая энцефалопатия и необратимые изменения внутренних органов, обычно приводящие к когнитивным расстройствам. Прочитайте дополнительные сведения уровни АЛК в моче могут быть повышены. В таких случаях следует определить уровень свинца в сыворотке. Если уровни АЛК и ПБГ в моче нормальные или только немного увеличены, для дифференциальной диагностики острых порфировых синдромов целесообразно измерить общее количество порфиринов в моче и определить профили этих порфиринов методом высокоэффективной жидкостной хроматографии.
Справочные материалы касающиеся вторичной порфиринурии
1. An G, Wang X, Morris ME: Flavonoids are inhibitors of human organic anion transporter 1 (OAT1)-mediated transport. Drug Metab Dispos 42(9):1357–1366, 2014. doi: 10.1124/dmd.114.059337
2. Duan P, Li S, Ni A, et al: Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: Discovery and molecular characterization. Mol Pharm 9(11):3340–3346, 2012. doi: 10.1021/mp300365t
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Поздняя кожная порфирия ( Урокопропорфирия , Хроническая печеночная порфирия )
Поздняя кожная порфирия – хроническое заболевание, характеризующееся повышенным образованием порфиринов и их накоплением в коже. При данной форме поражаются кожные покровы, подвергающиеся воздействию солнечного света (фотосенсибилизации). Патология проявляется гиперпигментацией участков кожи, повышенной ранимостью, гипертрихозом, образованием пузырей, эрозий и язв. Диагноз ставится на основании клинической картины, анамнеза, высокого содержания порфиринов в плазме, моче и кале. В качестве лечения проводятся сеансы плазмафереза и кровопусканий, назначаются аминохинолиновые и комплексообразующие препараты, солнцезащитные средства.
МКБ-10
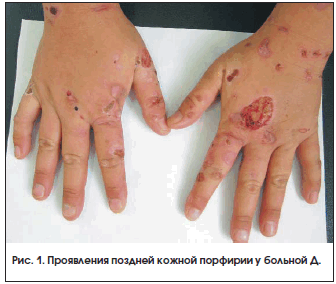
Общие сведения
Поздняя кожная порфирия (ПКП, урокопропорфирия, хроническая печеночная порфирия) относится к группе заболеваний с нарушением порфиринового метаболизма (порфирий) и является самой распространенной из них. Средняя частота встречаемости урокопропорфирии составляет 1:10000 человек, а в странах Западной Европы и Южной Африки ‒ 1:1000 и 1:800 человек соответственно. Манифестация ПКП наступает в возрасте 40-50 лет. Болеют преимущественно мужчины, на долю которых приходится 90-93% всех клинических случаев. Данная патология характеризуется хроническим течением с рецидивами в весенне-летний период, что связано с увеличением длины дня и солнечной активности.
Причины ПКП
В основе развития заболевания лежит недостаточная активность уропорфириноген-декарбоксилазы, что ведет к накоплению порфиринов, в больших концентрациях оказывающих токсическое действие. Порфирины ‒ органические тетрапиррольные соединения, которые синтезируются главным образом в печени и костном мозге и необходимы для образования гемоглобина, миоглобина, каталаз, пероксидаз, цитохрома Р-450 и витамина В12. Сниженная функция уропорфириноген-декарбоксилазы приводит к торможению синтеза порфиринов на уровне уропорфирина и копропорфирина.
Это может произойти вследствие двух механизмов. Первый – мутация гена А1В18, кодирующего фермент, второй – действие различных факторов, оказывающих ингибирующее действие на фермент. К таким факторам относятся гепатит С, алкоголизм, хроническая интоксикация тяжелыми металлами (мышьяк, свинец, ртуть), прием пероральных контрацептивов, длительный контакт с нефтепродуктами. Наиболее частая причина ПКП у мужчин – алкоголизм и гепатит С, у женщин – гормональные контрацептивы. Более редкими причинами являются ВИЧ-инфекция, саркоидоз, злокачественная опухоль печени и терминальная стадия хронической почечной недостаточности. Наличие наследственной и приобретенной форм – главная отличительная особенность ПКП от других нарушений порфиринового обмена, которые являются только наследственными патологиями.
Патогенез
В результате сниженной функции уропорфириноген-декарбоксилазы увеличивается концентрация копропорфирина и уропорфирина. Данные вещества, накапливаясь в кожных покровах, под действием длинноволнового спектра солнечных лучей индуцируют образование свободных радикалов, что запускает процессы перекисного окисления липидов и высвобождение протеолитических ферментов из тучных клеток. Это приводит к клеточному повреждению базальной мембраны эпидермиса и сосудов дермы, что обусловливает отслойку наружного слоя кожи и появление симптомов фотосенсибилизации (легкая ранимость, пузыри, эрозии, язвы).
Отложение порфиринов в коже, а также стимуляция образования меланина меланоцитами способствует усилению пигментации. Гиперпигментация и фототоксические реакции также связаны с высоким содержанием железа в крови. Нарушение метаболизма железа является частой сопутствующей патологией ПКП, так как развивается вследствие хронических заболеваний печени. Механизм ускоренного роста волос на лице (гипертрихоза) до сих пор неизвестен. Поражение нервной системы ограничивается дисфункцией ее вегетативного отдела и нейротрофическими нарушениями. При гистопатологическом исследовании кожи больных ПКП обнаруживается ее субэпидермальная отслойка, разрыхление рогового слоя эпидермиса, отделение эпидермиса от соединительнотканной части, характерная фестончатость сосочков дермы и утолщение эндотелия поверхностных кожных сосудов.
Классификация
В зависимости от преобладания тех или иных клинических проявлений различают кожную, кожно-нервную, кожно-висцеральную и смешанную формы. По степени выраженности симптомов разделяют манифестную и латентную ПКП. По этиологическому фактору выделяют следующие виды урокопропорфирии:
- Наследственная. Развивается вследствие генетической мутации. Наследование происходит по аутосомно-доминантному типу. Снижение уровня уропорфириноген-декарбоксилазы наблюдается в печени, эритроцитах, плазме.
- Спорадическая (приобретенная). Наиболее частая разновидность ПКП. Основные причины – алкоголизм, гепатит С, прием эстрогенов. Низкое содержание уропорфириноген-декарбоксилазы отмечается только в печени.
- Паранеопластическая. Возникает в результате развития гепатомы, продуцирующей избыточные количества порфиринов. Отличительной особенностью является поздний дебют (60-70 лет).
- Псевдо-ПКП. Развивается у больных хронической почечной недостаточностью в терминальной стадии, т. е. находящихся на гемодиализе. Клиническая картина возникает не по причине повышенной продукции порфиринов, а их сниженного выведения. Особенности данной формы – нормальная концентрация порфиринов в моче и очень высокая в плазме, выраженные фототоксические реакции.
Симптомы ПКП
Основной орган поражения – кожа. Наиболее типичными проявлениями манифестной формы считаются гиперпигментация, образование пузырей, гипертрихоз. Усиление пигментации наблюдается на участках кожи, систематически подвергающихся инсоляции (лицо, уши, шея, верхняя часть груди, кисти рук). Кожные покровы приобретают различные оттенки – от землисто-серого до бронзового. Интенсивность пигментации более выражена у брюнетов. В начале заболевания изменение цвета кожи носит временный характер, исчезающий в зимний период. По мере прогрессирования ПКП гиперпигментация становится постоянной. У некоторых пациентов долгое время это может быть единственным симптомом.
К признакам фоточувствительности относится повышенная ранимость кожи. Отслойка эпидермиса происходит даже при незначительных механических воздействиях. На коже образуются плотные пузыри размером до 1 см с жидкостным содержимым, которое может иметь серозный, гнойный или геморрагический характер. Через некоторое время пузыри вскрываются, оставляя после себя болезненные эрозии. После заживления эрозий формируются рубцы.
Для латентной ПКП характерны гипертрихоз височно-скуловой области лица, утолщение кожи на задней поверхности шеи с глубокими ромбовидными складками. Больные выглядят старше своих лет из-за появления или усиления морщин на лице. Вегетативно-трофические нарушения включают повышенную потливость, головные боли, нарушения сна, диарею или запор, учащение сердцебиения, усиление сосудистого рисунка на коже верхней части груди за счет пареза поверхностных капилляров (симптом «зарева» или «лимонной кожи»), деформацию ногтей, атрофию мышц кистей рук, предплечий, плечевого пояса.
Существуют атипичные варианты кожной порфирии. Язвенно-некротическая форма развивается у людей с ослабленным иммунитетом (сахарный диабет, ВИЧ-инфекция). При данном виде ПКП возникают глубокие язвенные дефекты кожи и гнойно-некротический распад мягких тканей. Инфильтративно-бляшечная, склеродермоподобная и витилигинозная формы имитируют изменения кожи при дискоидной красной волчанке (чешуйчатая эритема на лице в виде бабочки), диффузной склеродермии (уплотнение, кальциноз кожи) и витилиго (депигментированные пятна), что становится причиной частых диагностических ошибок.
Кожная порфирия у женщин имеет некоторые особенности течения: более раннее начало заболевания (около 30 лет), ограниченная пигментация на лице (хлоазмы), выраженный гипертрихоз, поражение нетипичных участков кожи – спина, бедра, подошвенная поверхность стоп. Среди женщин чаще, чем среди мужчин, встречаются витилигинозная, склеродермоподобная и инфильтративно-бляшечная формы ПКП.
Осложнения
Поздняя кожная порфирия ухудшает течение сердечно-сосудистых заболеваний, увеличивает риск развития сахарного диабета. Постоянные эрозии и язвенные дефекты кожных покровов часто осложняются бактериальной инфекцией. В редких случаях при язвенно-некротической форме развивается синдром системной воспалительной реакции и сепсис. Основную опасность для жизни представляют заболевания, которые вызывают ПКП: вирусный гепатит С и алкогольный гепатит, которые могут привести к циррозу и печеночной недостаточности, онкологические заболевания, хроническая почечная недостаточность, ВИЧ-инфекция.
Диагностика
Поздняя кожная порфирия представляет собой междисциплинарную проблему. В ее диагностике принимают участие врачи разных специальностей - дерматологи, гематологи, инфекционисты, гепатологи, ревматологи, онкологи. Так как в большинстве случаев кожная порфирия носит приобретенный характер, необходимо выяснить, на фоне какого заболевания она развилась. У пациента необходимо узнать об употреблении алкоголя, приеме оральных контрацептивов, наличии вирусного гепатита С. Для постановки диагноза проводятся следующие методы исследования:
- Рутинные лабораторные тесты. В биохимическом анализе крови отмечается увеличение печеночных трансаминаз, билирубина, гамма-глутамилтранспептидазы (специфичного показателя алкогольного поражения печени), ферритина, сывороточного железа, креатинина (при почечной недостаточности), снижение уровня альбумина. При циррозе печени в коагулограмме выявляются признаки гипокоагуляции. У многих пациентов обнаруживаются положительные маркеры вируса гепатита С.
- Специфические лабораторные исследования. Наблюдается повышение концентрации уропорфирина в плазме и моче, копропорфирина в кале. Под лампой Вуда моча принимает красно-оранжевое или ярко-розовое свечение. При люминесцентной микроскопии плазма дает красную флуоресценцию. Снижение активности фермента уропорфириноген-декарбоксилазы в эритроцитах и наличие генетической мутации характерно только для наследственной формы заболевания.
- Инструментальные исследования. Визуализирующие методы (ультразвуковое исследование и КТ брюшной полости) необходимо проводить для исключения злокачественного образования печени, продуцирующего порфирины. Это в первую очередь касается пациентов, у которых симптомы заболевания развились после 60 лет.
Кожную порфирию следует дифференцировать с другими видами порфирий, протекающими с поражением кожных покровов (наследственная копропорфирия, врожденная эритропоэтическая и вариегатная порфирии). Также ПКП дифференцируют с дерматологическими (фотодерматозы, вульгарная пузырчатка, герпетиформный дерматит Дюринга), ревматологическими заболеваниями (дискоидная красная волчанка, диффузная склеродермия), гемохроматозом, надпочечниковой недостаточностью.
Лечение ПКП
Особое внимание при лечении ПКП уделяется устранению факторов и патологий, которые привели к развитию заболевания. Это подразумевает полный отказ от алкоголя, прекращение приема оральных контрацептивов, лечение гепатита С, проведение антиретровирусной терапии, химиотерапии и хирургического удаления злокачественной опухоли печени.
Патогенетическое лечение ПКП включает в себя несколько мероприятий. С целью удаления избытка порфиринов и железа из крови проводятся сеансы флеботомий (кровопусканий) и плазмафереза. В дополнение к этим процедурам, а также при противопоказаниях к ним используют лекарственные средства, связывающие и выводящие порфирины и железо. К ним относятся синтетические противомалярийные препараты из группы аминохинолинов (хлорохин, гидроксихлорохин) и комплексообразующие соединения (дефероксамин, Д-пеницилламин). При применении аминохинолинов очень важно начинать их прием с малых доз. Это связано с тем, что обычные дозы в начале лечения вызывают специфическую токсическую реакцию (порфириновый криз), характеризующуюся усиленным синтезом порфиринов и резким ухудшением состояния пациента (повышение температуры тела, тошнота, рвота, боли в животе). Для уменьшения абсорбции порфиринов в желудочно-кишечном тракте назначаются энтеросорбенты (активированный уголь).
Для защиты кожных покровов от солнечного света рекомендуется ношение максимально закрытой одежды, использование солнцезащитных кремов и мазей, содержащих парсол- 1789 или мексомил ‒ вещества, не пропускающие ультрафиолетовые лучи длинноволнового спектра. Обязательна постоянная обработка эрозивных поверхностей кожи антисептическими растворами во избежание инфекционных осложнений.
Прогноз и профилактика
Среди всех патологий с нарушением обмена порфиринов кожная порфирия является наиболее благоприятной в плане течения и прогноза. Серьезные осложнения возникают редко, их причиной в основном являются заболевания или патологии, которые приводят к развитию ПКП. Профилактика заключается в избегании факторов, способствующих возникновению кожной порфирии (употребление алкоголя, прием оральных контрацептивов, длительное пребывание на солнце, контакт с тяжелыми металлами, нефтепродуктами). Людям, у кого в семье есть больной наследственной формой ПКП, показано определение активности уропорфириноген-декарбоксилазы и проведение молекулярно-генетической диагностики.
2. Дерматовенерология. Национальное руководство/ под ред. Ю.С. Бутова, Ю.К. Скрипкина, О.Л. Иванова. — 2016.
3. Поздняя кожная порфирия/ Оркин В.Ф., Шабогина А.А., Олехнович Н.М., Кочнева Е.В.// Клиническая дерматология и венерология. 2015. № 3.
4. Поздняя кожная порфирия/ Монахов С.А.// Российский журнал кожных и венерических болезней – 2014 -№ 5.
Порфирия ( Порфириновая болезнь )
Порфирии ‒ большая группа наследственных заболеваний, характеризующихся нарушением биосинтеза гема и накоплением его токсичных метаболитов. Клинические проявления крайне разнообразны – от светочувствительности и кожных высыпаний до болей в животе, полного паралича и острых психозов. Диагностика осуществляется с помощью молекулярно-генетических тестов, специальных лабораторных методов определения порфиринов и их предшественников в моче и кале, оценки активности ферментов в крови. Лечение заключается в мероприятиях, направленных на снижение образования токсических метаболитов, их выведение из крови, проведении симптоматической терапии и хирургических вмешательств.
Порфирии (от греч. «porphyreis» - пурпурный) – ряд заболеваний обмена веществ, при которых нарушается образование гема, в результате чего в организме накапливаются порфирины или их токсичные предшественники. Патологии данной группы встречаются относительно редко ‒ от 7 до 12 случаев на 100 000 человек. Отдельные нозологии имеют свою эндемичность. Так, распространенность поздней кожной порфирии в странах Южной Африки составляет 1:800, острой перемежающейся порфирии в Швеции ‒ 1:1000, вариегатной порфирии в Южной Африке ‒ 1:3000. У большинства порфирий нет гендерных различий, кроме поздней кожной формы (чаще страдают мужчины) и острой интермиттирующей (чаще болеют женщины).
Причины порфирий
В подавляющем большинстве случаев причиной порфирий выступают генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в биосинтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается вследствие заболеваний печени (алкогольный гепатит, вирусный гепатит С) или длительной интоксикации тяжелыми металлами. Наследование порфирий происходит по аутосомно-доминантному или аутосомно-рецессивному типу. Синтезирование гема протекает в 8 последовательных этапов, за каждый отвечает свой фермент, кодируемый определенным геном. Для каждой формы порфирии существует специфичный ферментативный дефект.
Гем представляет собой комплексное соединение порфиринов с двухвалентным железом. Наибольшее количество гема образуется в печени и костном мозге. В печени гем входит в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и обезвреживании различных ксенобиотиков. В костном мозге гем используется для образования гемоглобина. Результатом сниженной активности ферментов является торможение синтеза гема на определенном уровне, что ведет к накоплению его токсичных промежуточных метаболитов.
Помимо генетической мутации, для развития острых порфирий необходимо воздействие провоцирующих факторов, стимулирующих выработку порфиринов. Такими факторами являются голодание, длительная инсоляция, стрессы, алкоголь, инфекции, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства, подвергающиеся метаболизму системой цитохрома P-450 (нестероидные противовоспалительные препараты, антибиотики, антиконвульсанты, оральные контрацептивы, седативные средства). Особую роль играют колебания женских половых гормонов во время менструаций или беременности. У женщин месячные являются наиболее частым провоцирующим фактором, а беременность ассоциируется с тяжелым течением заболевания.
В результате неполноценности ферментов, участвующих в образовании гема, и действия провоцирующих факторов происходит увеличение концентрации его токсичных продуктов обмена. Для хронических порфирий характерно накопление протопорфирина, копропорфирина и упопорфирина. При острых формах возрастает количество порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Порфирины накапливаются в коже и под действием ультрафиолетового излучения (солнечного света) запускают процесс перекисного окисления липидов, вызывая деструкцию и гибель клеток кожи. Копропорфирин и протопорфирин усиливают пигментацию кожи и ускоряют рост волос (гипертрихоз). Плохо растворимый в воде протопорфирин откладывается в клетках печени, закупоривает портальные тракты и желчные протоки. Отложение уропорфирина в эритроцитах приводит к их ускоренному разрушению в селезенке (гемолиз). Предшественники порфиринов (ДАЛК и порфобилиноген), накапливаясь в нервной ткани, вызывают демиелинизацию и аксональную дегенерацию нервных волокон.
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность. Наиболее целесообразно выделять следующие виды порфирий:
- Эритропоэтические. К ним относятся врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера), эритропоэтическая протопорфирия (ЭПП). Основной клинический признак – поражение участков кожи, которые подвергаются воздействию прямых солнечных лучей (фотосенсибилизация). Данные патологии являются наиболее тяжелыми и имеют самый высокий процент летальности.
- Острые печеночные. Сюда включены острая перемежающаяся порфирия (ОПП), вариегатная порфирия (ВП), наследственная копропорфирия (НКП). Острые порфирии характеризуются приступообразным течением. Преимущественно страдает нервная система. При ВП и НКП также встречаются признаки фотосенсибилизации.
- Хронические печеночные. К ним относят позднюю кожную порфирию (ПКП), которая имеет наследственную и приобретенную формы. Это наиболее благоприятный вид порфирии.
Симптомы порфирий
Спектр клинических проявлений очень широк. Порфирии могут протекать в виде острых атак или хронически. Различия наблюдаются также в возрасте дебюта заболевания. Так, эритропоэтические порфирии манифестируют уже в дошкольном детстве (3-5 лет), острые порфирии - после полового созревания (14-16 лет), а спорадическая (приобретенная) форма ПКП - после 40 лет.
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
При поздней кожной форме возникает гиперпигментация участков кожи, подвергающихся постоянному воздействию солнечного света (лицо, шея, ушные раковины, верхняя часть груди, кисти рук). Кожа приобретает землистый или бронзовый оттенок. Также характерны гипертрихоз лобно-височной области лица, фотосенсибилизация, проявляющаяся повышенной ранимостью кожи и образованием пузырей с жидкостным содержимым. После вскрытия пузырей формируются эрозии. На местах разрешения эрозий образуются атрофические рубцы.
При эритропоэтических порфириях наблюдаются более выраженные признаки светочувствительности, чем при ПКП (ранимость, пузыри, эрозии). При длительном нахождении на свету появляется покраснение и сильное жжение кожи. Обширные эрозии оставляют после себя грубые рубцы на лице, что приводит к обезображиванию внешнего вида больного. В результате множественных рубцов на коже кистей рук развиваются контрактуры суставов, что значительно затрудняет их движения. Моча становится красной или розовой, а зубы окрашиваются в красно-коричневый цвет (эритродонтия). Из-за увеличенной селезенки могут появиться тяжесть или ноющие боли в левом подреберье. Специфический признак ЭПП – утолщение, огрубение и уплотнение кожи вокруг рта и глаз, на крыльях и спинке носа, на тыльных поверхностях кистей.
Нарушения порфиринового обмена ухудшают течение сердечно-сосудистых заболеваний, неблагоприятно влияют на углеводный метаболизм и повышают риск развития сахарного диабета 2 типа. Острые формы порфирий вследствие выраженной полинейропатии осложняются параличом дыхательной мускулатуры, аспирационной пневмонией, отеком головного мозга, тромбоэмболиями, рабдомиолизом. Постоянные эрозии кожных покровов могут привести к бактериальным инфекциям. При ЭПП из-за отложения нерастворимого в воде протопорфирина может развиться цирроз печени и печеночная недостаточность.
При подозрении на порфирию пациента направляют к врачу-гематологу. При постановке диагноза учитывается наличие заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, менструации, беременность). Лабораторная диагностика порфирий следующая:
- Клинико-биохимические анализы. При болезни Гюнтера в общем и биохимическом анализах крови выявляются признаки гемолиза (пойкилоцитоз, анизоцитоз, сфероцитоз, ретикулоцитоз, повышение непрямого билирубина и сывороточного железа), увеличение печеночных трансаминаз. У больных с острыми порфириями отмечается снижение уровня глюкозы и натрия, при ПКП - увеличение сывороточного железа и ферритина. Также у 80% с ПКП выявляются положительные маркеры вируса гепатита С.
- Специфические исследования. Для диагностики острых порфирий широко используется скрининговая проба Эрлиха (при смешивании специального реактива с мочой она окрашивается в красный цвет). Для ОПП характерно повышение ДАЛК и порфобилиногена в моче, для ВП - протопорфирина в кале, для НКП – копропорфирина в кале и моче. При ПКП в моче увеличено содержание уропорфирина, в кале – копропорфирина. При ЭПП наблюдается высокая концентрация протопорфирина в эритроцитах и кале, при ВЭП - уропорфирина в моче, кале и эритроцитах. При люминесцентной микроскопии плазма дает красное флуоресцирование при ПКП и эритропоэтических порфириях.
Также для подтверждения диагноза проводится определение уровня ферментов цикла биосинтеза гема в эритроцитах, лимфоцитах или плазме - порфобилиногендезаминазы (ОПП), копропорфириноген-оксидазы (НКП), протопорфириноген-оксидазы (ВП), уропорфириногенсинтетазы (ВЭП), уропорфириногендекарбоксилазы (ПКП), феррохелатазы (ЭПП). Заключительным этапом диагностики является молекулярно-генетическое тестирование для выявления мутаций генов, кодирующих перечисленные выше ферменты. Данные исследования особенно эффективны для распознавания асимптомных форм порфирий.
Эритропоэтические порфирии дифференцируют с дерматологическими заболеваниями (буллезным пемфигоидом, вульгарной пузырчаткой), с гематологическими патологиями, протекающими со спленомегалией (лейкозами, лимфомами, аутоиммунными гемолитическими анемиями) с болезнями почек. ПКП дифференцируют с заболеваниями печени, гемохроматозом, надпочечниковой недостаточностью. Острые порфирии следует дифференцировать с хирургическими заболеваниями, сопровождающимися сильной болью в животе, неврологическими и психиатрическими патологиями.
Лечение порфирий
Пациентов с острыми и эритропоэтическими порфириями необходимо госпитализировать отделение гематологии. Лечение ПКП возможно как в стационаре, так и в амбулаторных условиях. На сегодняшний день не существует эффективных методов, полностью ликвидирующих нарушения обмена порфиринов. Основной упор делается на патогенетическую и симптоматическую терапию, а также на устранение провоцирующих факторов. Способы лечения зависят от вида порфирий:
- Острые. Для подавления образования порфобилиногена и ДАЛК применяют гем-аргинат, производные АТФ (аденил, рибоксин) и большие дозы глюкозы с дальнейшим переходом на высокоуглеводную диету. Для купирования вегетативной симптоматики используют октреотид, для ускорения восстановления миелина в нервных волокнах - витамины группы В. При менструалозависимых атаках эффективна овариосупрессивная терапия. С этой целью назначают агонисты гонадотропин-рилизинг гормона.
- Эритропоэтические. Данные порфирии очень плохо поддаются терапии. Основное лечение – это защита кожных покровов от солнечного света (окна со стеклом, не пропускающим ультрафиолет, закрытая одежда, фотозащитные кремы, прием бета-каротина). Необходимо обрабатывать эрозии антисептическими растворами для профилактики инфекционных осложнений. При выраженном гемолизе показана спленэктомия. В некоторых случаях болезни Гюнтера эффективной оказывается трансплантация костного мозга. При ЭПП дополнительно назначаются гепатопротекторы (урсодезоксихолевая кислота, адеметионин) и антицирротическая терапия.
- ПКП. С целью удаления порфиринов и избытка железа проводят плазмаферез и флеботомию (кровопускания). При противопоказаниях к данным процедурам назначаются аминохинолиновые (хлорохин) и комплексообразующие (дефероксамин) препараты. Для уменьшения всасывания порфиринов в желудочно-кишечном тракте применяют энтеросорбенты (активированный уголь). Также используются солнцезащитные кремы. При наличии гепатита С необходима противовирусная терапия интерфероном-альфа и рибавирином.
В большинстве случаев порфирии являются тяжелыми заболеваниями с неблагоприятным прогнозом. При эритропоэтических формах продолжительность жизни составляет около 30 лет, смерть наступает от интеркуррентных инфекций. ЭПП часто приводит к циррозу печени. При атаках острых порфирий летальный исход наблюдается в 15-20%, основная причина смерти – паралич дыхательных мышц. При ПКП прогноз благоприятный, тяжелых осложнений не происходит. Для предупреждения рецидивов рекомендуется избегать провоцирующих факторов – инфекций, голодания, стрессов, длительной инсоляции, употребления алкоголя и определенных лекарственных средств. Людям, у которых в семье есть больной порфирией, необходимо определять активность ферментов цикла синтеза гема и проводить ДНК-диагностику для выявления генетических мутаций.
1. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена/ Кривошеев Б.Н. и др. – 2014.
2. Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. - 2018.
3. Диагностическая роль отдельных синдромов и симптомов в семиотике острых порфирий/ Пустовойт Я.С., Галстян Г.М., Савченко В.Г.//Гематология и трансфузиология. – 2014 - №59(3).
Диагностика и лечение острых порфирий
НАЦИОНАЛЬНЫЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
ДИАГНОСТИКА И ЛЕЧЕНИЕ ОСТРЫХ ПОРФИРИЙ
Москва 2018 г.
Порфирии представляют собой группу заболеваний, состоящую из семи нозологических форм. Причинами их возникновения являются генетически обусловленные нарушения активности различных ферментов в цепи биосинтеза гема (Рисунок 1, Приложения). Это приводит к нарушению обмена порфиринов, имеющему свои особенности при каждой форме порфирии и определяющему их клинические проявления. Начало изучения нарушений порфиринового обмена относится к 1841г., когда Scherer доказал, что красный цвет мочи больных обусловлен наличием в ней определѐнных пигментов, а не присутствием молекул гемоглобина. Fisher в 1930г. получил Нобелевскую премию за работу по изучению промежуточных продуктов гема и в 1934 году опубликовал книгу «Химия пирролов» [6]. Схема биосинтеза гема была описана в 50-е годы прошлого столетия.
В дальнейшем были идентифицированы все восемь ферментов в этом цикле [5]. Установлено, что каждая нозологическая форма порфирий связана с дефектом активности одного из ферментов (кроме синтетазы дельта-аминолевулиновой кислоты (-АЛК)), закодированного в одном гене.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
13-15 октября, Алматы, "Атакент"
600 брендов, более 150 компаний-участников из 20 стран.
Новинки рынка стоматологии. Цены от производителей
Порфирии подразделяются на эритропоэтические и печѐночные в зависимости от ткани, где происходит преимущественное нарушение метаболизма порфиринов (см. классификация I) [10]. Вместе с тем, порфирии могут подразделяться на формы с поражением кожных покровов и острые, провоцируемые формы (см. классификация II).
- Поздняя кожная порфирия
Острые формы порфирий характеризуются яркой неврологической и вегетативно-сосудистой симптоматикой, в основе которой лежит полинейропатия. При порфирии, обусловленной дефицитом дегидратазы δ-аминолевулиновой кислоты и острой перемежающейся порфирии (ОПП) нет кожных проявлений. Это объясняется тем, что у них активными метаболитами являются предшественники порфиринов (δ-АЛК и порфобилиноген (ПБГ)), которые не имеют сродства к тканям дермы.
При снижении активности ферментов поздних этапов биосинтеза гема происходит накопление собственно порфиринов, избыток которых в дерме приводит к фототоксическим реакциям. Вследствие этого клиника кожных поражений характерна для таких острых порфирий как: наследственная копропорфирия (НКП) и вариегатная порфирия (ВП), а также поздней кожной порфирии (ПКП) и эритропоэтических порфирий.
Этиология и патогенез
Развитие различных форм порфирий связано с нарушениями цикла биосинтеза гема и имеет общие черты. В основе развития каждой формы порфирии лежит генетически обусловленное снижение или отсутствие активности определѐнного фермента в цепи биосинтеза гема (рисунок). Гены ферментов расположены на разных хромосомах и не имеют групповой сцепленности. Снижение активности фермента до 50% от нормы может не иметь клинических проявлений.
При ОП реализовать генетическое носительство и спровоцировать клиническую манифестацию заболевания могут:
-лекарственные препараты (НПВС, барбитураты, цефалоспорины сульфаниламиды и др. список которых представлен в приложении)
Перечисленные факторы приводят к повышенному потреблению конечного продукта цикла биосинтеза – гема (например, активация системы цитохрома Р-450) [10], либо оказывают непосредственное стимулирующее воздействие на активность первого фермента цикла биосинтеза – синтетазы -АЛК, что приводит к повышению еѐ активности (например действие прогестерона) [7], в результате чего ускоряется синтез всех промежуточных продуктов метаболизма порфиринов. На этапе участия дефектного фермента начинается избыточное накопление метаболитов в токсических концентрациях, что приводит к обострению заболевания. При ОП избыточное накопление -АЛК и ПБГ в тканях приводит к сегментарной демиелинизации нервных волокон с нарушением нервной проводимости. Токсическому воздействию подвержены все отделы нервной системы человека.
Периферическая сенсорно-моторная полинейропатия является следствием вторичной демиелинизации нервных волокон.
Вовлечение вегетативной нервной системы имеет следующий патогенез: - поражение абдоминальных вегетативных сплетений сопровождается спазмом сосудов брыжейки и нарушением моторики кишечника.
- ослабление активности n. vagus приводит к преимущественному влиянию на сердечно-сосудистую систему симпатического отдела; этому также способствует 10-ти кратное увеличением экскреции катехоламинов и нарушение функции барорецепторов артериальных сосудов.
Нарушение функции центральной нервной системы является следствием токсического воздействия предшественников порфиринов на нейроны головного мозга и развития длительного спазма артериол, гипонатриемии и гипергидратации, что приводит к тяжѐлым энцефалопатиям.
Поздняя кожная порфирия (ПКП) и эритропоэтические порфирии имеют хроническое течение с периодами обострений, которые могут вызвать:
Повышенная светочувствительность кожных покровов связана с фотохимическими реакциями, спровоцированными порфиринами. Избыток порфиринов в коже подвергается активному воздействию спектра солнечного излучения с длинами волн 400 – 410 нм, что приводит к образованию реактивных частиц, например, супероксид аниона, активирующего ксантин-оксидазу, и других метаболитов, повреждающих клетки базальной мембраны. Повторные атаки приводят к развитию нескольких слоѐв базальных мембран и образованию пласта кровеносных сосудов в поверхностных слоях дермы. Реактивные кислородсодержащие частицы также могут приводить к высвобождению гистамина из тучных клеток, усиливая явления фототоксичности.
Изменения кожи. Уропорфириноген – основной метаболит при поздней кожной порфирии стимулирует синтез фибробластами коллагена. При эритропоэтической протопорфирии жирорастворимый протопорфириноген откладывается в стенке сосудов дермы, приводя к их утолщению.
Пигментирование кожи и гипертрихоз наблюдаются в периорбитальных областях при поздней кожной порфирии и эритропоэтических порфириях, однако, механизм этих изменений до конца не изучен.
Эпидемиология
Порфирии не являются эндемичными заболеваниями и с одинаковой частотой встречаются среди населения всех континентов. Частота встречаемости острых форм порфирий (ОП) по различным оценкам составляет 7-12 случаев на 100 тысяч здоровых людей. В то же время частота бессимптомного носительства генетических дефектов, приводящих к ОП, составляет ~ 50-100 случаев на 100000 человек.
Клиническая картина
Cимптомы, течение
-эритема, волдыри на открытых участках кожи.
Первый приступ острых порфирий может развиться в возрасте старше 14-16 лет, значительно чаще у женщин. Начало заболевания острое, реже подострое. После воздействия порфириногенных факторов появляются боли в животе, конечностях, пояснице, тошнота, рвота. К концу второй недели заболевания появляется мышечная слабость, переходящая в парезы и параличи. Характерны тахикардия (до 110-130 артериальная гипертензия (до 180/100), выделение мочи с красноватым оттенком, неадекватное поведение и галлюцинации. При отсутствии лечения в течение 20-40 дней у больных могут развиться бульбарные нарушения и паралич дыхательной мускулатуры. В анализах крови нередко выявляется гипонатриемия.
Лабораторная диагностика острых порфирий, кроме порфирии обусловленной дефицитом дегидратазы --АЛК основывается на определении в моче избытка порфобилиногена с помощью:
- качественного скрининг-теста свежего образца мочи больного с использованием реактива Эрлиха по методу Watson-Schwartz. При наличии в моче избытка ПБГ образуется окрашенный продукт розово-красного цвета;
- количественного определения содержания ПБГ в моче (норма не превышает 2 мг/л).
При порфирии, обусловленной дефицитом дегидратазы-АЛК в моче, определяется высокая концентрация дельта-аминолевулиновой кислоты при нормальном содержании ПБГ.
Диагноз острой порфирии устанавливается на основании характерной клинической картины и высокого содержания ПБГ или -АЛК в моче [1,17].
Дифференциальный диагноз между ОПП и ВП или НКП основывается при измерении содержания общих порфиринов в кале [1,17]. В норме концентрация порфиринов в кале менее 200 нмоль/г сухого веса. Нормальная концентрация общих порфиринов в кале подтверждает диагноз ОПП. Повышение концентрации в несколько раз свидетельствует в пользу НКП или ВП. При исследовании плазмы спектрофлюориметрическим методом можно дифференцировать НКП от ВП.
Следующим этапом диагностики является определение активности ферментов в клетках крови. Это позволяет подтвердить диагноз у больных с клиническими проявлениями заболеваний и выявить бессимптомных носителей. Несмотря на распространѐнность и удобство диагностики бессимптомного носительства порфирий путѐм оценки активности специфического фермента в клетках, этот метод не является абсолютно достоверным [8,6].
Поэтому заключительным этапом диагностики порфирий у больных и, в особенности, у бессимптомных носителей является проведение ДНК- анализа [2].
Причиной поражения кожных покровов может быть повышенная светочувствительность, как следствие одной из форм порфирий. Тем не менее, следует провести дифференциальную диагностику кожной порфирии с другими дерматологическими заболеваниями.
- определение общих порфиринов в плазме и исследование спектра ее поглощения при флюоресцентной спектроскопии;
Для подтверждения диагноза ЭПП следует провести дополнительный тест - определение общих порфиринов в эритроцитах. При повышенной их концентрации следует измерить соотношение свободного и Zn-связанного протопорфиринов. В случае значительного превышения концентрации свободного протопорфирина подтверждается диагноз ЭПП.
Если результаты всех перечисленных тестов у пациента с активными кожными нарушениями будут нормальными, то диагноз порфирии будет маловероятным..
В случае же получения хотя бы одного или нескольких положительных результатов подтверждается диагноз кожной порфирии, форма которой определяется с помощью диагностических тестов.
Дифференциальный диагноз
- соматизация и синдром хронической усталости
Первичный этап диагностики, на практике, является самым сложным и ответственным для врача. Вариативность течения и симптоматики ОП (особенно при атипичном и моносимптомном вариантах развития) создают значительные трудности в своевременной постановке этого диагноза, нередко уводя врача в сторону ошибочных предположений. Выполнение же скрининг теста на наличие избытка ПБГ в моче всем обращающимся без анализа первичных жалоб нерационально с точки зрения временных и материальных затрат. Опытным путѐм, подтверждѐнным статистическими данными, выделены характерные для ранних сроков течения и специфичные для ОП симптомы. Разработана шкала, содержащая данные анамнеза и симптомы как характерные для клинического течения острых порфирий, так и наиболее часто ложно приписываемых ОП. Скрининг имеющихся у пациентов симптомов и сопоставление этих симптомов с симптоматикой, представленной в таблице (приложение 1)*, позволяет по сумме баллов судить о степени вероятности наличия порфирии у больного. Такой подход позволяет минимизировать число ятрогений, связанных с назначением пациентам с неустановленным диагнозом порфириногенных препаратов.
*В шкале представлены симптомы, встречающиеся при острых порфириях, которым присвоено определенное количество баллов, имеющих как положительные, так и отрицательные значения в зависимости от частоты их встречаемости при острой порфирии. Эти баллы суммируются, и если сумма баллов составляет менее 5 - диагноз острой порфирии маловероятен, если от 5 до 15 баллов – диагноз порфирии возможен, если более 15 баллов, то диагноз острой порфирии высоко вероятен.
Поражение кожных покровов при порфириях следует дифференцировать с прочими дерматологическими заболеваниями.
Лечение
1. Острые порфирии, цель лечения – предупреждение атак заболевания и развития необратимых изменений нервной системы.
Профилактика развития приступов ОП предусматривает ограничение воздействия на организм провоцирующих факторов, а именно:
- приѐма лекарственных препаратов с повышенной порфириногенной активностью, инсоляции развития бактериальных и вирусных инфекции (особенно HCV, HBV, CMV), состояния гипогликемии
- у женщин с доказанной связью между наступлением menses и частыми приступами – предупреждение менструаций.
Если менструальные циклы часто (три и более раз в год) провоцируют атаки ОП, репродуктивную функцию необходимо подавлять, для чего используют оральные контрацептивы – Ригевидон, Овидон; ГнРГ- Золадекс, синарел; андрогены - сустанон, андриол; овариоэктомия в отдельных случаях. Отмена овариосупрессии производится после достижения длительного бесприступного (несколько месяцев и более) течения заболевания.
Лечение начинают при наличии нескольких симптомов ОП (см. выше) и повышении показателей порфиринового обмена (по сравнению с предыдущими данными в динамике). При развитии острого приступа необходимо немедленное начало патогенетической терапии для подавления избыточного биосинтеза порфиринов:
2. Обеспечение избыточного поступления в орагнизм углеводов (200-600гр сухого вещ-ва глюкозы). 40%-1000мл в/в капельно в сутки, ежедневно, 2-4 недели [4].
3. Сандостатин в дозе 100-500 мкг/сут., подкожно, ежедневно на протяжении от 4-х недель до 6 месяцев в сочетании с плазмаферезами 6-10 сеансов [13].
4. Рибоксин 2%-10мл в разведении на 100-200 мл 0.9% раствора NaCl, в/в капельно 1-2 раза в сутки, ежедневно 2-4 недели.
Читайте также:
- Лечение аутоиммунного тиреоидита. Лекарства и операция
- Врожденная холестеатома вершины пирамиды височной кости - лучевая диагностика
- Клиника гнойного менингита. Неврологические симптомы гнойного менингита. Тактика при гнойном менингите.
- Лучевая диагностика зубовидной кости
- Извращенный аппетит (пикацизм) и его причины