Пероксисомальные расстройства
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Эпилепсия — заболевание головного мозга, соответствующее любому из следующих состояний:
- Не менее двух неспровоцированных (или рефлекторных) эпилептических приступов с интервалом > 24 ч.
- Один неспровоцированный (или рефлекторный) эпилептический приступ и вероятность повторных приступов, соответствующая общему риску рецидива (³ 60 %) после двух неспровоцированных эпилептических приступов, в следующие 10 лет.
- Диагноз эпилептического синдрома.
Критерии разрешения эпилепсии включают достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом либо отсутствие эпилептических приступов в течение 10 лет у пациентов, не получавших противосудорожные препараты более 5 лет.
Классификация эпилепсии (Международная противоэпилептическая лига (ILAE) 2017 г. )
Классификация эпилепсии проводится после определения критериев диагностики эпилепсии (определение выше). Классификация проводится с использованием трехуровнего ранжирования — определение типа приступов, типа эпилепсии и синдрома эпилепсии. Нейроимиджинг, ЭЭГ и другие исследования, если они есть, помогают улучшить классификацию на всех трех уровнях. Где это возможно, следует установить диагноз на всех трех уровнях. Этиологию эпилепсии следует устанавливать с самого начала и на каждом этапе всего диагностического пути. Знание этиологии может способствовать оптимизации классификации и имеет важные лечебные последствия для пациента.
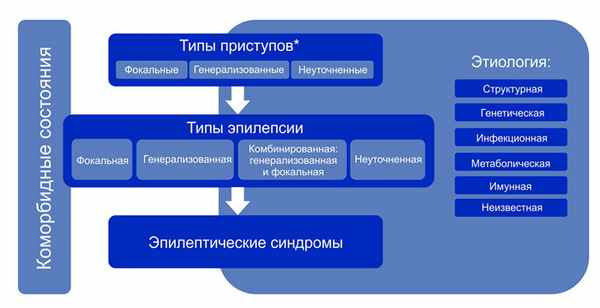
Структура Классификации эпилепсии ILAE 2017 г.
Примечание. * Оценивается по началу приступа.
Авакян Г. Н. Блинов Д. В. Лебедева А. В. Бурд С. Г. Авакян Г. Г. Классификация эпилепсии Международной Противоэпилептической Лиги: пересмотр и обновление 2017 года. Эпилепсия и пароксизмальные состояния. 2017; 9 (1): 6–25. DOI: 10.17749/2077–8333.2017.9. 1.006–025.
Алгоритм классификации эпилепсии:
- На первом этапе (уровне) определяют тип приступа: фокальный, генерализованный или с неизвестным началом.
- На втором этапе (уровне) устанавливают тип эпилепсии: фокальная, генерализованная или сочетанная фокальная и генерализованная, или неизвестная (unknown).
- На третьем этапе (уровне) определяют эпилептический синдром и коморбидность. Эпилептический синдром представляет собой совокупность характеристик, включая тип приступа, данные ЭЭГ и нейровизуализации, часто имеет возрастзависимый характер, провоцирующие факторы, хронозависимость и в ряде случаев определенный прогноз.
- На четвертом этапе (уровне) устанавливают этиологию эпилепсии: структурная, генетическая, инфекционная, метаболическая, иммунная или с неизвестной этиологией.
Генерализованная эпилепсия
Пациенты с генерализованной эпилепсией имеют генерализованные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ паттерны, которые сопровождают генерализованные типы приступов (например, генерализованный спайк-волна). Сопутствуют семейная история генерализованных типов приступов или генерализованная эпилепсия.
Генетическая / идиопатическая генерализованная эпилепсия представляет собой эпилепсию с генерализованными приступами, ассоциированнами с генерализованными эпилептиформными паттернами ЭЭГ, такими как генерализованная спайк-волновая активность, которая, считается, имеет генетическую этиологию. Однако это не всегда означает, что эти эпилепсии унаследованы или могут передаваться потомству, поскольку генетическая этиология может быть спонтанной новой мутацией или наследование может быть сложным. Таким образом, термин «идиопатическая генерализованная эпилепсия» используется как синоним генетической генерализованной эпилепсии, и клиницист может выбрать, какой термин использовать, в зависимости от важности акцента на генетическом наследовании для конкретного пациента. Генетические / идиопатические генерализованные эпилепсии включают детскую абсансную эпилепсию, юношескую абсансную эпилепсию, юношескую миоклоническую эпилепсию, и эпилепсию с генерализованными тонико-клоническими приступами.
Фокальная эпилепсия
Пациенты с фокальной эпилепсией имеют фокальные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ-паттерны, которые сопровождают фокальные типы приступов (такие как фокальные острые волны или фокальное интериктальное замедление). Нейроимиджинг демонстрирует фокальную структурную аномалию мозга и способствует установке диагноза, хотя пациенты с генетической этиологией и нормальной визуализацией могут также иметь фокальную эпилепсию. Фокальные эпилепсии могут быть унифокальными, мультифокальными или полушарными.
Сочетанная генерализованная и фокальная эпилепсия
Пациенты могут иметь как генерализованные, так и фокальные типы приступов, с интериктальными и / или иктальными ЭЭГ-паттернами, которые сопровождают оба типа приступов. Пациенты с синдромом Драве и синдромом Леннокс-Гасто могут иметь генерализованную и фокальную эпилепсию.
Неизвестная эпилепсия
Термин «неизвестный» используется для обозначения в случае, когда понимается, что у пациента есть эпилепсия, но невозможно определить, является ли она фокальной, генерализованной или комбинированной: фокальной и генерализованной. Это бывает при недостаточной информации для классификации эпилепсии, например, если ЭЭГ является нормальной / неинформативной.
Синдромы эпилепсии
В то время как концептуализация эпилепсий по их этиологии очень важна, эпилепсии также могут быть организованы (в соответствии с установленными достоверными общепринятыми клиническими и ЭЭГ — характеристиками) в эпилептические синдромы. Такие синдромы имеют типичный возраст начала приступа, специфические типы приступов и характеристики ЭЭГ и часто другие признаки, которые вместе взятые позволяют диагностировать конкретный эпилептический синдром. Идентификация синдрома эпилепсии полезна, так как она предоставляет информацию о том, какие основные этиологии следует учитывать и какие антиконвульсанты могут быть наиболее полезными. Некоторые эпилептические синдромы демонстрируют аггравацию приступов при определенных антиконвульсантах, чего можно избежать при ранней диагностике синдрома эпилепсии.
Эпилептические синдромы
Неонатальный и младенческий период:
- самокупирующиеся неонатальные приступы и самокупирующаяся семейная неонатальная эпилепсия;
- самокупирующаяся семейная и несемейная младенческая эпилепсия;
- ранняя миоклоническая энцефалопатия;
- синдром Отахара;
- синдром Веста;
- синдром Драве;
- миоклоническая эпилепсия младенчества, младенческая эпилепсия с мигрирующими фокальными приступами;
- миоклоническая энцефалопатия при непрогрессирующих заболеваниях;
- фебрильные приступы, фебрильные приступы плюс, генетическая эпилепсия с фебрильными приступами плюс.
Детский возраст:
- эпилепсия с миоклонически-атоническими (ранее – астатическими) приступами;
- синдром Леннокса–Гасто;
- фебрильные приступы, фебрильные приступы плюс;
- абсансная эпилепсия;
- эпилепсия с миоклоническими абсансами;
- детская затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса);
- детская затылочная эпилепсия с поздним дебютом (синдром Гасто);
- фотосенситивная затылочная эпилепсия;
- самокупирующаяся эпилепсия с центрально-темпоральными спайками;
- атипичная эпилепсия с центрально-темпоральными спайками;
- эпилептическая энцефалопатия с продолженной пик-волновой активностью во время сна;
- синдром Ландау–Клеффнера;
- аутосомно-доминантная ночная лобная эпилепсия.
Подростковый и взрослый возраст:
- юношеская абсансная эпилепсия;
- юношеская миоклоническая эпилепсия;
- эпилепсия с изолированными генерализованными тонико-клоническими приступами;
- аутосомно-доминантная эпилепсия со слуховыми проявлениями;
- другие семейные височные эпилепсии.
Любой возраст:
- семейная фокальная эпилепсия с вариабельным фокусом;
- рефлекторные эпилепсии;
- прогрессирующие миоклонические эпилепсии.
Этиология эпилепсии
В последние годы значительно расширилось наше понимание основополагающих причин эпилепсии, подкрепленное достижениями в области современной нейровизуализации и генетического тестирования. Такая терминология, как «идиопатическая», «криптогенная» и «симптоматическая», больше не используется. Эпилепсии теперь описываются более точно конкретными основополагающими причинами.
Генетическая этиология
Понятие генетической эпилепсии заключается в том, что эпилепсия, насколько мы понимаем, является прямым результатом известного или предполагаемого генетического дефекта (ов), в котором судороги являются основным симптомом расстройства. Генетический дефект может возникать на хромосомном или молекулярном уровне. Важно подчеркнуть, что «генетический» не означает то же, что «унаследовано», поскольку мутации de novo не являются редкостью. Наличие генетической этиологии не исключает экзогенного влияния на возникновение эпилепсии.
Наиболее важные генетические причины эпилепсии, которые могут быть идентифицированы при клиническом тестировании:
Существует много способов, которыми генетические факторы могут способствовать развитию эпилепсии. Определенные генетические факторы, возможно, не были унаследованы и не могут быть переданы потомству. Вот некоторые важные генетические концепции, используемые на этом веб-сайте, и их определения:
- унаследованные аномалии генов, аутосомно-доминантное, аутосомно-рецессивное и менделевское наследование;
- приобретенные аномалии генов — de novo, спорадические, мозаицистические, зародышевые и соматические;
- полигенная / комплексная генетическая этиология.
Структурная этиология
Структурные эпилепсии определяются как имеющие выраженную структурную аномалию мозга, которая связана с существенно повышенным риском эпилепсии. Структурная аномалия мозга может быть приобретена (например, вследствие инсульта, травмы или инфекции) или может быть генетического происхождения; однако, как мы это понимаем в настоящее время, структурная аномалия мозга представляет собой отдельное нарушение, расположенное между приобретенным или генетическим дефектом и эпилепсией.
Нейроимиджинг
Магнитно-резонансная томография (МРТ) с использованием 1.5 Тесла аппарата является минимальным стандартным исследованием для исключения структурной аномалии. При этом исследовании важно выполнять протоколы, специфические для эпилепсии, которые позволяют тщательно изучать специфические приобретенные аномалии (например, склероз гиппокампа ) и тонкие пороки развития коры головного мозга, такие как фокальная дисплазия коры. Изображение с использованием 3 Тесла и использование передовых методов анализа программного обеспечения может помочь в выявлении структурных нарушений, не очевидных при обычной МРТ. Интериктальная и иктальная ЭЭГ вместе с дополнительными функциональными исследованиями нейровизуализации, такими как ПЭТ, ОФЭКТ и МЭГ, помогают внимательно изучить конкретную область мозга и идентифицировать тонкую аномалию. У детей младшего возраста в возрасте до 2 лет тонкие аномалии не могут быть выявлены из-за незаконченной миелинизации, и повторное исследование требуется в динамике.
Общие структурные аномалии мозга, связанные с эпилепсией:
- пороки развития коры головного мозга;
- сосудистые пороки развития;
- гиппокампальный склероз;
- гипоксически-ишемические структурные аномалии;
- травматическая повреждение мозга;
- опухоли;
- порэнцефалическая киста.
Метаболическая этиология
Метаболические эпилепсии определяются как имеющие определенное метаболическое нарушение, связанное с выраженным риском развития эпилепсии. Метаболические расстройства имеют генетическое происхождение; однако, как мы это понимаем в настоящее время, метаболические аномалии представляют собой отдельное нарушение, стоящее между генетическим дефектом и эпилепсией.
Важные метаболические эпилепсии:
- дефицит биотинидазы и голокарбоксилазы-синтазы;
- дефицит церебрального фолата;
- нарушения креатина;
- приступы при нарушении фолатного цикла;
- недостаточность транспортера глюкозы 1 (GLUT1);
- митохондриальные расстройства;
- пероксисомальные расстройства;
- пиридоксинзависимая эпилепсия.
Иммунная этиология
Иммунные эпилепсии определены как имеющие выраженную иммунную опосредованную этиологию с подтверждением воспаления центральной нервной системы, что, как было показано, связано с существенно повышенным риском развития эпилепсии.
Важные иммуноопосредованные эпилепсии:
- Синдром Расмуссена;
- Антителоопосредованная эпилепсия.
Инфекционная этиология
Наиболее распространенная этиология эпилепсии во всем мире является инфекционной, особенно в развивающихся странах. Инфекции в центральной нервной системе могут вызывать как острые симптоматические припадки (которые тесно связаны со сроками первичной инфекции), так и эпилепсией. Инфекционная этиология включает туберкулез, ВИЧ, церебральную малярию, нейроцистицеркоз, подострый склерозирующий панэнцефалит, церебральный токсоплазмоз. Эти инфекции иногда имеют структурный коррелят, однако основная причина эпилепсии определяется как инфекционный процесс. Инфекционная этиология может иметь специфические последствия лечения. Существуют также последствия для общественного здравоохранения, поскольку профилактика таких инфекций может снизить нагрузку на эпилепсию, особенно в развивающихся странах. Наиболее распространенные из таких инфекций следующие:
- бактериальный менингит или менингоэнцефалит;
- церебральная малярия;
- церебральный токсоплазмоз;
- цитомегаловирусная инфекция;
- ВИЧ;
- нейроцистицеркоз;
- туберкулез;
- вирусный энцефалит;
- подострый склерозирующий панэнцефалит;
- прочие инфекции (токсокариоз, шистосомоз, болезнь Лайма (нейроборрелиоз).
Неизвестная этиология
«Неизвестная» этиология должна рассматриваться нейтрально и обозначать, что природа основной причины возникновения эпилепсии пока неизвестна; это может быть фундаментальный генетический дефект или отдельное, пока еще установленное, нарушение.
Имитаторы эпилепсии
Существует ряд состояний, связанных с рецидивирующими пароксизмальными событиями, которые могут имитировать симптомы, и ошибочно диагностироваться как эпилепсия. Важно, чтобы эти расстройства учитывались при оценке пароксизмальных событий, так как частота ошибочных диагнозов при эпилепсии высока во всем мире. История заболевания и видеозапись приступов помогают установить правильный диагноз. Существуют некоторые состояния, при которых могут сосуществовать эпилептические и неэпилептические события.
Пероксисомальные расстройства
Для получения дополнительной информации см. таблицу Расстройства биогенеза пероксисом и метаболизма очень длинноцепочечных жирных кислот .
Синдром Зельвегера (СЗ), неонатальная адренолейкодистрофия и инфантильная болезнь Рефсума (ИБР)
Эти нарушения являются тремя проявлениями продолжающегося заболевания, от наиболее (ZS) до наименее (IRD) тяжелого. Вызывающий заболевание генетический дефект возникает в 1 из по крайней мере 12 генов, участвующих в формировании пероксисом или импорте протеинов (PEX-семейство генов).
Проявления включают дисморфизм лица, пороки развития центральной нервной системы, демиелинизацию, неонатальные судороги, гипотонию, гепатомегалию, кистоз почек, короткие конечности с шероховатым эпифизом (точечная хондродисплазия), катаракту, ретинопатию, дефицит слуха, задержку психомоторного развития и периферическую нейропатию.
Диагноз подозревают на основании повышенного содержания в крови жирных кислот с очень длинной цепью, фитановой кислоты, промежуточных продуктов синтеза желчных кислот и пипеколиновой кислоты; для подтверждения диагноза проводят генетическое тестирование.
На данный момент специфического лечения этих заболеваний нет. Ведение заболевания в основном симптоматическое.
Ризомелиевая точечная хондродисплазия
Этот дефект пероксисомального биогенеза вызван мутациями гена PEX7 и характеризуется скелетными изменениями, включающими гипоплазию средней части лица, поразительно короткие проксимальные конечности, выступающие лобные бугры, маленькие ноздри, катаракту, ихтиоз и глубокую задержку психомоторного развития. Позвоночные расщелины также распространены.
Диагноз точечной дисплазии ризомелического типа предполагают на основании результатов рентгенографического исследования, возрастания уровня фитановой кислоты в сыворотке крови и низких уровней эритроцитарного плазмалогена в сыворотке крови; уровни VLCFA в норме. Подтверждение осуществляется при помощи генетического тестирования.
Нет специфического лечения точечной дисплазии ризомелического типа.
Х-сцепленная адренолейкодистрофия
Это расстройство вызвано недостатком пероксисомального мембранного переносчика ALDP, кодируемого геном ABCD1. Этот ген связан с Х-хромосомой и, таким образом, заболевание проявляется главным образом у мужчин. В настоящее время идентифицировано > 900 мутаций (см. ALD Info).
Церебральная форма поражает 40% больных. Начало происходит между 4 и 8 годами, и симптомы дефицита внимания Клинические проявления СДВГ является синдромом невнимательности, гиперактивности и импульсивности. Выделяют 3 типа СДВГ: преимущественно невнимательный, преимущественно гиперактивный/импульсивный и комбинированный. Прочитайте дополнительные сведения прогрессируют с течением времени до серьезных поведенческих нарушений, слабоумия, и нарушений зрения, слуха и моторных функций, что приводит к полной нетрудоспособности и смерти в течение 2-3 лет после установления диагноза. Также были описаны более мягкие подростковые и взрослые формы.
Около 45% больных имеют более мягкую форму, которая называется адреномиелонейропатией (АНМ); начало заболевания в возрасте 20–30 лет, с прогрессирующим парапарезом и нарушением сфинктера и половой функции. Около трети этих пациентов также имеют церебральные симптомы.
У пациентов с любой формой может также развиться недостаточность надпочечников Болезнь Аддисона Аддисонова болезнь – это медленно развивающаяся и обычно прогрессирующая гипофункция коры надпочечников. Она сопровождается различными симптомами, включая артериальную гипотонию и гиперпигментацию. Прочитайте дополнительные сведенияДиагноз X-сцепленной адренолейкодистрофии предполагается на основании изолированного повышения ОДЦЖК и подтверждается на основании секвенирования генов.
Трансплантация костного мозга или стволовых клеток Трансплантация гемопоэтических стволовых клеток Трансплантация гемопоэтических стволовых клеток (ГСК) – быстроразвивающаяся технология, которая потенциально может позволить добиться излечения при злокачественных заболеваниях крови ( лейкемиях. Прочитайте дополнительные сведения может помочь в некоторых случаях стабилизировать симптомы. Заместительная терапия стероидами надпочечников необходима для пациентов с надпочечниковой недостаточностью. Добавление в рацион смеси 4:1 глицерина триолеата и глицерина триеруката (масло Лоренцо) может нормализовать плазменные уровни VLCFA,но, по-видимому, не останавливает неврологическую дегенерацию у симптоматичных пациентов. Однако если давать его мальчикам до появления симптомов, это может замедлить прогрессирование заболевания; однозначная польза не была определена. В настоящее время проводятся клинические испытания в области генной терапии, которые уже оказались весьма успешными.
Классическая болезнь Рефсума
Генетический дефицит одного пероксисомного фермента – фитаноил-КоA-гидроксилазы, катализирующей метаболизм фитановой кислоты (общий компонент растительной пищи), – приводит к накоплению фитановой кислоты.
Клинические проявления включают прогрессирующую периферическую нейропатию, нарушение зрения, обусловленное пигментным ретинитом Пигментная дегенерация сетчатки Пигментная дистрофия сетчатки – это медленно прогрессирующая двухсторонняя дегенерация сетчатки и ее пигментного эпителия, вызываемая различными генетическими мутациями. Клиническими проявлениями. Прочитайте дополнительные сведенияДиагноз болезни Рефсума подтверждается повышением фитановой кислоты и снижением уровня Пристановой кислоты в сыворотке (повышение фитановой кислоты сопровождается повышением Пристановой кислоты в ряде других пероксисомальных нарушений).
Лечение болезни Рефсума – диетическое ограничение фитановой кислоты ( < 10 мг/день), которое может быть эффективным для предотвращения или задержки симптомов, если начать его до появления симптомов.
Дополнительная информация
Ниже следуют англоязычные ресурсы, которые могут быть информативными. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов.
ALD Info (адренолейкодистрофия): ресурс, предоставляющий информацию о лечении, диагностике и других аспектах адренолейкодистрофии
Online Mendelian Inheritance in Man® (OMIM®) database: полная информация о генах и их молекулярной и хромосомной локализации
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Отделение детской и подростковой медицины ( 229507 )

Об отделении | Университетская клиника Гамбург-Эппендорф
Отделение детской и подростковой медицины при университетской клинике Гамбург-Эппендорф предлагает полный спектр услуг по диагностике и лечению различных заболеваний у новорожденных, младенцев, маленьких детей и подростков. Отличительной чертой отделения является уникальная компетенция в области лечения крайне редких и сложных патологий у маленьких пациентов. Здесь ежегодно проходят лечение более 17000 амбулаторных и около 4600 стационарных пациентов. Кроме того, при отделении ведет работу дневной стационар (около 1000 пациентов в год). Главврач отделения – профессор, доктор медицины Аня Мунтау.
Все врачи и сестринский персонал отделения отлично ладят с детьми и пытаются установить доверительные отношения с маленьким пациентом. Отделение заслуженно гордится не только ультрасовременной технической базой, но и прекрасной инфраструктурой с игровыми комнатами для детей.
Основные направления клинической деятельности отделения охватывают:
- Диагностика и лечение дегенеративных заболеваний головного мозга/деменции у детей
- Нейрональный цероидный липофусциноз
- Лейкодистрофия
- Дегенеративные заболевания головного мозга неясного генеза
- Эпилепсия (медикаментозная терапия, стимуляция блуждающего нерва, кетогенная диета и т.д.)
- Мышечные заболевания (сертификация Немецкого общества по мышечным заболеваниям)
- Туберозный склероз головного мозга
- Хроническая почечная недостаточность и заместительная почечная терапия (перитонеальный диализ, гемодиализ, трансплантация почки)
- Острая почечная недостаточность (включая процедуры гемофильтрации и плазмафереза)
- Врожденные и приобретенные гломерулопатии и тубулопатии
- Психологические консультации и консультации по питанию
- Диагностика в рамках специализированной лаборатории
- Поражения легких при ревматологических, онкологических, неврологических или метаболических заболеваниях
- Ювенильный идиопатический артрит
- Реактивный артрит
- Воспалительные ревматические системные заболевания (например, васкулиты, коллагенозы, дерматомиозит)
- Синдром периодической лихорадки
- Фенилкетонурия и другие нарушения метаболизма аминокислот
- Нарушения цикла метаболизма мочевины
- Ацидопатии
- Митохондриопатии
- Пероксисомальные расстройства
- Нарушения липидного обмена
Резюме Проф. Доктор мед. Аня Мунтау
Академическая и профессиональная деятельность
- С 2014 Заведующая отделением детской и подростковой медицины при университетской клинике Гамбург-Эппендорф.
- 2010 ‐ 2014 Заведующая отделением по лечению врожденных нарушений обмена веществ, детская клиника доктора фон Хаунершена, Мюнхенский университет, Мюнхен.
- 2007 - 2014 Руководитель проекта (LMU Excellence Initiative).
- 2006 - 2014 Профессор (W2) по педиатрии.
- 2004 ‐ 2014 Заведующая отделением педиатрии, детская клиника доктора фон Хаунершена, Мюнхенский университет, Мюнхен, а также руководитель проекта Баварской сети Центров по исследованию генома.
- 2001 - 2004 Постдокторант, детская клиника доктора фон Хаунершена, Мюнхенский университет, Мюнхен.
- 2000 - 2001 Постдокторант, Институт физиологической химии при Рурском университете в Бохуме.
- С 2000 Профессиональная сертификация в области детской и подростковой медицины.
- 1997 ‐ 2004 Подготовка по биохимической генетике и молекулярной биологии.
- 1990 ‐ 2000 Подготовка к профессиональной сертификации в области педиатрии.
- 1984 ‐ 1990 Изучение медицины человека в Мюнхенском университете имени Людвига-Максимилиана.
Награды и почетные членства
- С 2015 Член Национальной академии наук Леопольдина.
- 2012 Премия имени Терезы фон Байерн.
- 2008 Honorary Mention, премия за наставничество и науку (Nature Awards for Mentoring and Science).
- 2004 Премия имени Адельберта Черни за выдающиеся исследования в области педиатрии.
- 2001 - 2005 Исследовательская стипендия Немецкого научно-исследовательского общества.
Членство в профессиональных обществах
- Немецкое общество детской и подростковой медицины.
- Общество изучения врожденных нарушений обмена веществ.
- Американское общество генетики человека.
- Немецкая академия наук Леопольдина.
- Рабочая группа по нарушениям обмена веществ в детском возрасте.
Фото врача: (c) Universitätsklinikum Hamburg-Eppendorf (UKE)
Отделение детской и подростковой медицины.
Университетская клиника Гамбург-Эппендорф:спасибо за Ваш запрос.
В течении 1-го рабочего дня медицинский консультант изучит Ваш запрос и свяжется с Вами по телефону (высветится немецкий или Ваш локальный номер).
Пероксисома - Peroxisome - Wikipedia
А пероксисома (IPA: [pɛɜˈɹɒksɪˌsoʊm] ) [1] мембраносвязанный органелла (ранее известный как микротело ), обнаруженные в цитоплазме практически всех эукариотический клетки. [2] Пероксисомы - это окислительные органеллы. Часто молекулярный кислород служит дополнительным субстратом, из которого пероксид водорода (ЧАС2О2) затем формируется. Свое название пероксисомы получили благодаря деятельности по выработке и улавливанию перекиси водорода. Они играют ключевую роль в метаболизме липидов и превращении активные формы кислорода. Пероксисомы участвуют в катаболизм из жирные кислоты с очень длинной цепью, жирные кислоты с разветвленной цепью, промежуточные соединения желчных кислот (в печени), D-аминокислоты, и полиамины, то снижение из активные формы кислорода - конкретно пероксид водорода. [3] - и биосинтез плазмалогены, т.е. эфирные фосфолипиды важен для нормального функционирования мозга и легких млекопитающих [4] Они также содержат примерно 10% общей активности двух ферментов (Глюкозо-6-фосфатдегидрогеназа и 6-фосфоглюконатдегидрогеназа ) в пентозофосфатный путь [5] , что важно для энергетического обмена. [4] Активно обсуждается, участвуют ли пероксисомы в изопреноид и холестерин синтез у животных. [4] Другие известные пероксисомальные функции включают: глиоксилатный цикл в прорастающих семенах ("глиоксисомы "), фотодыхание в листьях, [6] гликолиз в трипаносомы ("гликосомы "), и метанол и / или окисление и ассимиляция амина в некоторых дрожжи.
Содержание
История
Пероксисомы (микротела) были впервые описаны шведским докторантом Дж. Родином в 1954 году. [7] Они были определены бельгийским цитологом как органеллы. Кристиан де Дюв в 1967 г. [8] Де Дюв и его сотрудники обнаружили, что пероксисомы содержат несколько оксидаз, участвующих в производстве перекиси водорода (H2О2) а также каталаза участвует в разложении H2О2 к кислороду и воде. Из-за их роли в метаболизме пероксидов Де Дюв назвал их «пероксисомами», заменив ранее использовавшийся морфологический термин «микротела». Позже было описано, что люцифераза светлячков нацелена на пероксисомы в клетках млекопитающих, что позволяет обнаружить сигнал нацеливания импорта для пероксисом и инициировать многие достижения в области биогенеза пероксисом. [9] [10] .
Структурный дизайн
Пероксисомы - это небольшие (диаметром 0,1–1 мкм) субклеточные компартменты (органеллы) с тонким зернистым матриксом, окруженные единственной биомембраной, которые расположены в цитоплазме клетки. [11] [12] Компартментализация создает оптимизированную среду для стимулирования различных метаболических реакций внутри пероксисом, необходимых для поддержания клеточных функций и жизнеспособности организма.
Количество, размер и белковый состав пероксисом варьируются и зависят от типа клеток и условий окружающей среды. Например, в дрожжах пекарских (С. cerevisiae ), было замечено, что при хорошем снабжении глюкозой присутствует только несколько маленьких пероксисом. Напротив, когда дрожжи были снабжены длинноцепочечными жирными кислотами в качестве единственного источника углерода, может образоваться от 20 до 25 больших пероксисом. [13]
Метаболические функции
Основная функция пероксисомы - расщепление жирные кислоты с очень длинной цепью через бета-окисление. В клетках животных длинные жирные кислоты превращаются в жирные кислоты со средней длиной цепи, которые впоследствии отправляются на митохондрии где они в конечном итоге распадаются на углекислый газ и воду. В клетках дрожжей и растений этот процесс осуществляется исключительно в пероксисомах. [14] [15]
Первые реакции образования плазмалоген в клетках животных также встречаются в пероксисомах. Плазмалоген - самый распространенный фосфолипид в миелин. Дефицит плазмалогенов вызывает серьезные нарушения миелинизации нервные клетки, что является одной из причин, почему многие пероксисомальные расстройства влияют на нервную систему. [14] Пероксисомы также играют роль в производстве желчь кислоты, важные для усвоения жиров и жирорастворимых витаминов, таких как витамины A и K. Кожные заболевания являются особенностями генетических нарушений, влияющих на функцию пероксисом в результате [15] .
Специфические метаболические пути, которые происходят исключительно в пероксисомах млекопитающих: [4]
- α-окисление фитановой кислоты
- β-окисление очень длинноцепочечных и полиненасыщенных жирных кислот
- биосинтез плазмалогенов
- конъюгация холевой кислоты как часть синтеза желчной кислоты
Пероксисомы содержат окислительные ферменты, Такие как Оксидаза D-аминокислот и оксидаза мочевой кислоты. [16] Однако последний фермент отсутствует у людей, что объясняет болезнь, известную как подагра, вызванный накоплением мочевой кислоты. Определенные ферменты внутри пероксисомы, используя молекулярный кислород, удаляют атомы водорода из определенных органических субстратов (обозначенных как R) в окислительной реакции, производя пероксид водорода (ЧАС2О2, сам токсичен):
Каталаза, другой фермент пероксисомы, использует этот H2О2 для окисления других субстратов, в том числе фенолы, муравьиная кислота, формальдегид, и алкоголь, посредством реакции перекисного окисления:
Эта реакция важна для клеток печени и почек, где пероксисомы выводят токсические вещества из организма, попадающие в кровь. Около 25% этиловый спирт что люди потребляют алкогольные напитки, окисляется до ацетальдегид таким образом. [14] Кроме того, при избытке H2О2 накапливается в клетке, каталаза превращает ее в H2О через эту реакцию:
У высших растений пероксисомы содержат также сложную батарею антиоксидантных ферментов, таких как супероксиддисмутаза, компоненты цикл аскорбат-глутатион и НАДФ-дегидрогеназы пентозофосфатного пути. Было продемонстрировано, что пероксисомы генерируют супероксид (O2 •− ) и оксид азота ( • НЕТ) радикалов. [17] [18]
Теперь есть доказательства того, что эти активные формы кислорода, включая пероксисомальный H2О2 также являются важными сигнальными молекулами у растений и животных и способствуют здоровому старению и возрастным нарушениям у людей. [19]
Пероксисома растительных клеток поляризуется при борьбе с проникновением грибов. Инфекция вызывает глюкозинолат Молекула, играющая противогрибковую роль, должна производиться и доставляться за пределы клетки под действием пероксисомальных белков (PEN2 и PEN3). [20]
Пероксисомы у млекопитающих и людей также способствуют противовирусной защите. [21] и борьба с патогенами [22]
Сборка пероксисом
Пероксисомы могут быть получены из гладкая эндоплазматическая сеть в определенных экспериментальных условиях и размножаются за счет роста мембраны и деления уже существующих органелл. [23] [24] [25] Белки матрикса пероксисомы транслируются в цитоплазму перед импортом. Конкретные аминокислотные последовательности (PTS или пероксисомальный целевой сигнал ) на C-конец (PTS1) или N-конец (PTS2) белков пероксисомального матрикса сигнализирует им, что они будут импортированы в органеллы, с помощью целевого фактора. В настоящее время известно 36 белков, участвующих в биогенезе и поддержании пероксисом, называемых пероксины [26] , которые участвуют в процессе сборки пероксисом у разных организмов. В клетках млекопитающих имеется 13 охарактеризованных пероксинов. В отличие от импорта белка в эндоплазматический ретикулум (ER) или митохондрии, белки не нужно разворачивать, чтобы импортировать в просвет пероксисомы. Рецепторы импорта белков матрикса, пероксины PEX5 и PEX7, сопровождают свой груз (содержащий аминокислотную последовательность PTS1 или PTS2 соответственно) до пероксисомы, где они высвобождают груз в пероксисомальный матрикс, а затем возвращаются в цитозоль - шаг с именем переработка отходов. Особый способ нацеливания на пероксисомальный белок называется «копилка». Белки, которые транспортируются этим уникальным методом, не имеют канонического PTS, а скорее связываются с белком PTS, который транспортируется в виде комплекса. [27] . Модель, описывающая цикл импорта, называется удлиненный челночный механизм. [28] Теперь есть доказательства того, что гидролиз АТФ необходим для рециркуляции рецепторов в цитозоль. Также, убиквитинирование имеет решающее значение для экспорта PEX5 из пероксисомы в цитозоль. Биогенез пероксисомальной мембраны и встраивание белков пероксисомальной мембраны (PMP) требует пероксинов PEX19, PEX3 и PEX16. PEX19 представляет собой рецептор PMP и шаперон, который связывает PMP и направляет их к пероксисомной мембране, где он взаимодействует с PEX3, интегральным мембранным белком пероксисомы. Затем PMP вставляются в пероксисомальную мембрану.
Разложение пероксисом называется пексофагией. [29]
Пероксисомное взаимодействие и общение
Разнообразные функции пероксисом требуют динамического взаимодействия и сотрудничества со многими органеллами, участвующими в клеточном метаболизме липидов, такими как эндоплазматический ретикулум (ER), митохондрии, липидные капли и лизосомы. [30]
Пероксисомы взаимодействуют с митохондриями в нескольких метаболических путях, включая β-окисление жирных кислот и метаболизм активных форм кислорода. [4] Обе органеллы находятся в тесном контакте с эндоплазматическим ретикулумом (ER) и разделяют несколько белков, включая факторы деления органелл. [31] Пероксисомы также взаимодействуют с эндоплазматическим ретикулумом (ER) и участвуют в синтезе эфирных липидов (плазмалогенов), которые важны для нервных клеток (см. Выше). У мицелиальных грибов пероксисомы перемещаются по микротрубочкам «автостопом» - процессом, включающим контакт с быстро движущимися ранними эндосомами. [32] Физический контакт между органеллами часто опосредуется участками контакта с мембраной, где мембраны двух органелл физически связаны, чтобы обеспечить быстрый перенос небольших молекул, обеспечить связь органелл и иметь решающее значение для координации клеточных функций и, следовательно, здоровья человека. [33] Изменения мембранных контактов наблюдаются при различных заболеваниях.
Сопутствующие медицинские условия
Пероксисомальные расстройства представляют собой класс заболеваний, которые обычно влияют на нервную систему человека, а также на многие другие системы органов. Два общих примера: Х-сцепленная адренолейкодистрофия и нарушения биогенеза пероксисом. [34] [35]
PEX гены кодируют белковые механизмы («пероксины»), необходимые для правильной сборки пероксисом, как описано выше. Для сборки и обслуживания мембраны требуются три из них (пероксины 3, 16 и 19), и они могут происходить без импорта ферментов матрикса (просвета). Разрастание органелл регулируется Pex11p.
Гены, кодирующие пероксиновые белки, включают: PEX1, PEX2 (PXMP3), PEX3, PEX5, PEX6, PEX7, PEX9 [36] [37] , PEX10, PEX11A, PEX11B, PEX11G, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26, PEX28, PEX30, и PEX31. У разных организмов нумерация и функции PEX могут различаться.
Эволюционное происхождение
Содержание белка в пероксисомах варьируется в зависимости от вида или организма, но наличие белков, общих для многих видов, было использовано для предположения эндосимбиотический источник; то есть пероксисомы произошли от бактерий, которые вторглись в более крупные клетки как паразиты, и очень постепенно развили симбиотические отношения. [38] Однако недавние открытия поставили под сомнение эту точку зрения. [39] Например, мутанты без пероксисом могут восстанавливать пероксисомы при введении гена дикого типа.
Два независимых эволюционных анализа пероксисомального протеом обнаружил гомологию между механизмом импорта пероксисом и ERAD путь в эндоплазматический ретикулум [40] [41] , наряду с рядом метаболических ферментов, которые, вероятно, были привлечены из митохондрии. [41] Недавно было высказано предположение, что пероксисома могла иметь актинобактериальный источник [42] Однако, это спорно. [43]
Пероксисома
Пероксисом является сотовая органелл окружена trilamellar мембраной и не содержит генетический материал или рибосомы. В отличие от митохондрий или хлоропластов, все составляющие его белки кодируются ядерными генами и происходят из цитозоля (или грубого эндоплазматического ретикулума для меньшинства трансмембранных белков). Пероксисома была открыта в 1965 году Кристианом де Дюве с помощью электронного микроскопа и разработки методов фракционирования клеток.
Пероксисомы участвуют в метаболизме жирных кислот и аминокислот, восстановлении реактивных производных кислорода и синтезе плазмалогенов .
Пероксисомы являются частью класса органелл, называемых «микротелами». Большинство авторов не рассматривают их как часть эндомембранной системы, поскольку они не участвуют в перманентном векторном потоке.
Резюме
Методы исследования
Мы можем приступить к разрыву и центрифугированию, которое разделит органеллы в зависимости от их плотности.
Также используется цитохимическое обнаружение, в частности, при исследовании каталазы. Таким образом, каталаза или уратоксидаза обнаруживается при контакте пероксисомы с субстратами этих белков. Благодаря иммуномаркированию золотыми шариками видно, что каталаза присутствует по всей пероксисоме, в то время как уратоксидаза специфична для паракристаллической зоны нуклеоида.
Можно также воспользоваться преимуществами специфичности определенных пероксисомальных белков и наблюдать за ними с помощью адресации GFP. Таким образом, зеленый флуоресцентный белок направляется в пероксисому, и его наблюдают под микроскопом, где области, где присутствует GFP, будут флуоресцентно помечены.
Структуры
Они представляют собой сферические органеллы размером от 0,1 до 1 мкм (называемые микропероксисомами или микротелами) до 1 мкм в диаметре у животных , но могут достигать 1,7 мкм у растений .
Они в основном состоят из пузырьков, соединенных канальцами, которые составляют матрикс или просвет пероксисомы, богатой белками.
Они окружены простой мембраной в виде липидного бислоя . Они присутствуют во всех эукариотических клетках (кроме ретикулоцитов ), с максимальным размером у животных в клетках печени ( гепатоцитах ) и почек .
Пероксисомы имеют нуклеоид, который представляет собой паракристаллическую область белка (уратоксидаза), за исключением приматов. Они содержат окисляющие ферменты: D-аминокислота-оксидаза, урат-оксидаза и каталаза . (для реакций, завершающих потребление O 2 ).
Кроме того, это не изолированные структуры, пероксисомы связаны между собой тонкими канальцами → сетью внутри цитоплазмы.
Протеин
Все белки, необходимые для пероксисом, синтезируются вне их, в цитозоле .
Мы находим в мембранных белках (негликозилированных), которые выполняют особую функцию по импорту ферментов, необходимых для функционирования пероксисомы.
С одной стороны, пероксины, отвечающие за систему импорта белка. Они кодируются генами PEX.
Сигналы адресации, распознаваемые пероксинами, - это PTS 1 и 2, соответственно, расположенные на стороне C и N.
Транспортеры ABC , использующие АТФ, отвечают за транспортировку (импорт и экспорт) метаболитов, присутствующих в мембране.
Цитохром P450 также специфичен для пероксисомы.
Функции
Как и митохондрии , пероксисомы являются важными участками утилизации кислорода O 2. ( реакции окисления ):
- Ферменты оксидазы (D-аминокислота-оксидаза, урат-оксидаза) удаляют свободные атомы водорода (реакция окисления ) со специфических органических субстратов R. Эти субстраты, связанные с атомами водорода , потенциально токсичны для клетки. Окисление этих молекул выводит их токсины.
RH 2 + O 2 → R + H 2 O 2 ; - каталаза использует перекись водорода Н 2 О 2 генерируется другими ферментами для окисления множества других токсичных субстратов R '(фенолов, метановой кислоты, спирта): это называется реакцией перекисного окисления. Этот тип реакции очень важен в печени , клетках почек, где пероксисомы выводят токсины, которые проходят через кровь .
Н 2 О 2 + R'H 2 ⤇ R '+ 2 H 2 O. Каталаза также катализирует реакцию: H 2 O 2 + H 2 O 2 ⤇ O 2 + 2H 2 O. Каталаза - самый распространенный фермент в пероксисомах; - если есть избыток H 2 O 2 , каталаза превращает его непосредственно в воду: 2 H 2 O 2 → 2 Н 2 О + О 2 . Это защитная реакция, потому что H 2 O 2 слишком много вредно для клетки;
- пероксисомы выполняют бета-окисление с длинной цепью жирных кислот с помощью механизма , подобных митохондрий . Пероксисомы даже обладают исключительным свойством этого пути у дрожжей и растений. Тем не менее, энергетический баланс сводится к производству ацетил-кофермента А, потому что электроны восстановленных коферментов приводят к образованию пероксида водорода, детоксицируемого на месте каталазой ;
- в листьях растений хлорофилла они участвуют в фотодыхании ;
- в клетках семян во время прорастания они связаны с липидными тельцами, из которых они позволяют образовывать углеводы, необходимые для роста проростка . В этом случае пероксисома получает название глиоксисомы .
- Реакция детоксикации: 50% выпитого алкоголя выводится в печени благодаря пероксисомам с образованием ацетальдегида, эта реакция катализируется каталазами .
Источник
Похоже, что эти органеллы возникли в результате бинарного расщепления родительских пероксисом. Однако недавно было замечено, что у больных людей, у которых больше нет пероксисом, исправление геномного происхождения болезни позволило появиться новым пероксисомам. Таким образом, происходит синтез пероксисом из гладкой эндоплазматической сети, называемой ранними пероксисомами .
Существуют автоматические циклы слияния и деления пероксисом. В этих циклах участвует DRP (родственный динамину белок), он вызывает сокращение мембраны. В ответ на определенные метаболические ситуации происходит умножение пероксисом.
В липидах , необходимые для производства новых пероксиса мембран также импортируется из цитоплазмы.
Согласно старой гипотезе, пероксисомы возникают в результате эндосимбиоза прокариота. Компартмент позволил бы, до митохондрий, снизить уровень O 2 (который является токсичным) в клетке. Эта гипотеза, вероятно, была опровергнута недавними результатами, которые скорее предполагают, что пероксисома происходит из эндоплазматического ретикулума.
Читайте также: