Пигментный дерматоз (синдром Блоха-Сульцбергера) - клиника, диагностика
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Синдром Блоха-Сульцбергера (Bloch-Sulzberger) - синонимы, авторы, клиника
Синонимы синдрома Блоха-Сульцбергера. Меланобластоз Bloch — Sulzberger. Пигментный дерматоз Siemens — Bloch. Недержание пигмента (incontinentia pigmenti). Семейный хроматоформный невус. Линейный или генерализованый меланобластоз кожи (Carol и Воиг). Пигментный дерматоз (Doornink). Дегенеративный меланоз дермы (Siemens). Системный пигментный невус (Siemens).
Определение синдрома Блоха-Сульцбергера. Редкий врожденный пигментный дерматоз (пигментная дисплазия). Относится к группе множественных пороков развития.
Авторы. Bloch Bruno — швейцарский дерматолог, 1878—1933. Sulzberger Marion Baldur — современный американский дерматолог. Впервые кожные проявления синдрома описал в 1925 г. Lechleuthner. В 1925 г. Bloch демонстрировал больного. Обобщающая работа Sulzberger была опубликована в 1928 г. Н. W. Siemens в 1929 г. сообщил о первом случае синдрома, который описал в 1925 г. Lechleuthner.
Симптоматология синдрома Блоха-Сульцбергера:
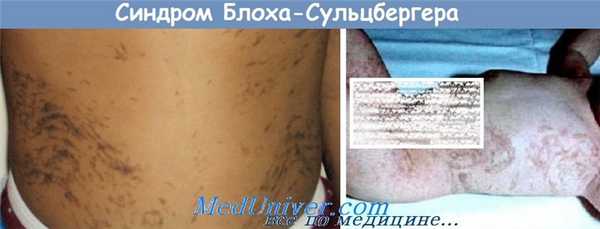
1. Чаще всего в грудном или раннем детском возрасте появляются грязновато-коричневатого или стального цвета очаги гиперпигментации кожи различной формы и размеров. Преимущественная локализация — боковые поверхности туловища; встречаются также симметричные очаги по передней и задней средней линии. На конечностях поражаются области плеч, а также бедер (до паховой области). Кожа лица, а также слизистые оболочки у большинства больных остаются свободными от высыпаний.
Первичными элементами сыпи иногда бывают буллезные и веррукозные, а также эритематозные и папуло-везикулярные эффлоресценции. Гиперпигментированные участки в течение ряда лет бледнеют и подвергаются инволюции с частичным исходом в атрофию.
2. Часто обнаруживаются и другие аномалии различного характера:
а) другие изменения кожи — алопеция, дистрофия ногтей, образование невусов;
б) поражения зубов — задержка развития и гипоплазия зубов, дефекты в их расположении, микродентия, гиподентия, шипообразные и выступающие вперед зубы;
в) поражения глаз — страбизм, псевдоглиома, врожденное помутнение роговицы и хрусталика, перемещение пигмента в роговице и сосудистой оболочке, атрофия зрительного нерва, микрофтальмия, синие склеры, птоз;
г) поражения центральной нервной системы — микроцефалия, дебильность, S. Little, эпилептиформные судороги, атаксия; д) гирсутизм;
е) врожденный вывих бедра.
3. Чаще заболевают женщины.
Этиология и патогенез синдрома Блоха-Сульцбергера. По-видимому, речь идет о наследственном заболевании, связанном с полом (передача поврежденной Х-хромосомы). Дерматоз относится к группе множественных пороков развития (эмбриопатий). Возможна связь с синдромом буллезного эпидермолиза.
Дифференциальный диагноз синдрома Блоха-Сульцбергера. Другие комбинированные пороки развития. Чешуйчатый лишай. S. Riehl. Токсическая меланодермия. Пойкилодермия. Парапсориаз. Пигментная крапивница (S. Nettleship). Красный плоский лишай. Пигментные невусы. S. Brandt. Синдром буллезного эпидермолиза. S. Naegeli I. S. Goltz — Gorlin.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Блоха-Сульцбергера
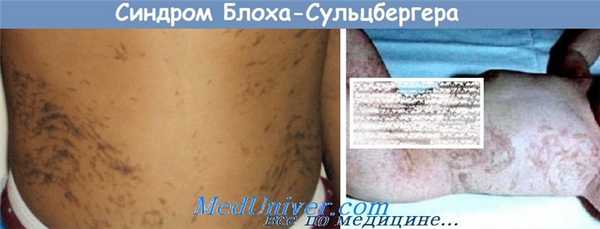
Синдром Блоха-Сульцбергера – наследственная форма нарушения пигментации кожи, которая часто сочетается с пороками развития зубов, волос, ногтей и глаз. Симптомы заболевания характеризуются выраженной стадийностью – сначала на коже появляется эритематозная сыпь в виде пятен и линий, затем на ее месте развивается гиперкератоз, сменяющийся пятнами и последующей гипопигментацией с атрофией кожных покровов. Диагностика синдрома Блоха-Сульцбергера производится на основании данных настоящего статуса больного, гистологического исследования образцов кожи в области поражения, изучения наследственного анамнеза и молекулярно-генетических анализов. Специфического лечения этой патологии на сегодняшний момент не существует, используют симптоматические и поддерживающие мероприятия различного характера.
Общие сведения
Синдром Блоха-Сульцбергера (семейная форма недержания пигмента, нейрокожный меланобластоз) – генетическое заболевание, характеризующееся нарушением метаболизма меланина в коже и рядом сопутствующих пороков развития. Впервые это заболевание было описано в 1926 году швейцарским дерматологом Б. Блохом, затем более детальное изучение данной патологии провел американский педиатр М. Сульцбергер в 1929 году и, независимо от предыдущих исследователей, немецкий врач Г. Сименс. Именно поэтому в литературе можно найти другое название этого заболевания – синдром Блоха-Сименса. Удалось выяснить, что патология наследуется сцепленно с Х-хромосомой, при этом мутантный аллель является доминантным. По этой причине синдром Блоха-Сульцбергера во много раз чаще встречается у девочек – половое распределение составляет примерно 6:210, так как наличие этой мутации у эмбрионов мужского пола практически всегда является летальным и приводит к самопроизвольному прерыванию беременности. Развитие заболевания у мальчиков может быть обусловлено генетическим мозаицизмом, наличием сопутствующего синдрома Клайнфельтера или редких точечных «мягких» мутаций. Общая встречаемость синдрома Блоха-Сульцбергера составляет примерно 1 случай на 75 000 новорожденных.
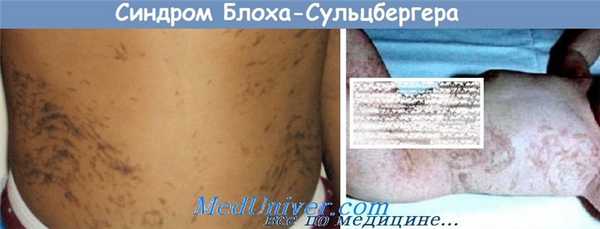
Причины синдрома Блоха-Сульцбергера
При синдроме Блоха-Сульцбергера происходит повреждение гена IKBKG, который располагается на Х-хромосоме. Продуктом его экспрессии является многофункциональный сложный белок – регуляторная субъединица NEMO-ингибиторной киназы, участвующей в сигнальной системе важного транскрипционного фактора (NF-каппа-B). Этот фактор и соответствующий ему сигнальный путь регулирует огромное количество различных процессов в организме человека – участвует в процессах адаптации при стрессе, иммунном ответе, некоторых формах воспалительных реакций, процессах клеточной адгезии, а также тормозит процессы апоптоза. Наиболее часто причиной синдрома Блоха-Сульцбергера становятся крупные транслокации и делеции гена IKBKG, в результате чего экспрессия и выделение белка с этого гена полностью прекращаются.
Поскольку у женщин имеется две Х-хромосомы, при наличии второй нормальной аллели гена IKBKG такая мутация не угрожает жизни, но обуславливает развитие синдрома Блоха-Сульцбергера. Известен факт, что в соматических клетках женского организма всегда активна только одна Х-хромосома, тогда как вторая сконденсирована в половой хроматин. Значительная вариабельность выраженности симптомов заболевания обусловлена распределением клеток, где активна хромосома именно с мутантной формой гена IKBKG. Как следствие, в вышеуказанных клетках не образуется регуляторной субъединицы NEMO-ингибиторной киназы, что и приводит к характерным порокам развития, формирующим клиническую картину синдрома Блоха-Сульцбергера. Кожные симптомы связаны с нарушением проницаемости мембран меланоцитов (в результате чего практически весь пигмент беспрепятственно покидает клетки) и аутоиммунными реакциями.
В отличие от женщин, у мужчин в норме есть лишь одна Х-хромосома, поэтому при наличии нонсенс-мутации в гене IKBKG выделение важного белка не происходит абсолютно во всех клетках организма. Это становится причиной массированного апоптоза гепатоцитов еще на этапе внутриутробного развития – в норме этот процесс задерживается как раз системой NF-каппа-B. Развитие нарушений, подобных синдрому Блоха-Сульцбергера, у мальчиков возможно при наличии сопутствующего синдрома Клайнфельтера (кариотипа XXY) или генетического мозаицизма, когда только часть клеток в организме имеет дефект гена IKBKG. В последние годы были выявлены точечные мутации этого гена, которые не приводят к полной остановке транскрипции, но изменяют структуру конечного белка. Однако чаще всего у мальчиков с такими дефектами возникает не синдром Блоха-Сульцбергера, а другие генетические заболевания – эктодермальные дисплазии, иммунодефициты, пороки развития скелета.
Симптомы синдрома Блоха-Сульцбергера
Одним из наиболее выраженных и распространенных проявлений синдрома Блоха-Сульцбергера является дерматоз, который обнаруживается при рождении или (реже) возникает на протяжении первых дней жизни новорожденного. В развитии изменений кожных покровов при этой патологии наблюдается характерная стадийность, что также является важным диагностическим признаком. Локализация таких изменений – на боковых поверхностях конечностей, туловища, шеи, вдоль линий Шарко или проекций основных нервных стволов. В большинстве случаев выделяется четыре основных стадии кожных симптомов синдрома Блоха-Сульцбергера:
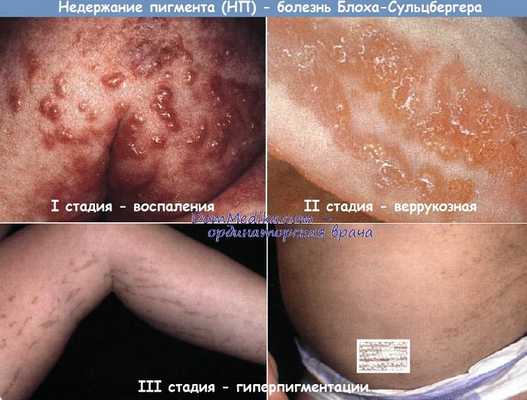
1-я стадия – воспалительная или везикулобуллезная. Начинается при рождении больного или на протяжении 2-3 недель жизни и длится до возраста 3-8 месяцев. На этом этапе заболевания возникают везикулы, эритематозная сыпь, возможно развитие пузырей и пустул. С учетом возраста больных синдромом Блоха-Сульцбергера существует определенный риск инфекционных осложнений на пораженных участках кожи.
2-я стадия – гипертрофическая или веррукозная. Характеризуется развитием на пораженных участках тела гиперкератоза в виде бляшек, бородавчатых и лихеноидных разрастаний. Их распределение, как правило, симметричное и линейное, вдоль линий Шарко или проекций нервных стволов. Длительность этой стадии синдрома Блоха-Сульцбергера составляет несколько месяцев (до возраста одного года), у некоторых больных может отсутствовать.
3-я стадия – пигментная. На этом этапе заболевания у больных на пораженных участках кожи возникают очаги гиперпигментации различных форм и размеров темно-коричневого цвета. Почти у половины пациентов с синдромом Блоха-Сульцбергера такие очаги появляются на неизмененных участках тела и не связаны с высыпаниями, характерными для предыдущих стадий. Длительность гиперпигментации составляет несколько лет, обычно – до периода полового созревания.
4-я стадия – атрофическая. Характеризуется потерей пигмента на очагах поражения с развитием признаков атрофии кожи. У некоторых больных синдромом Блоха-Сульцбергера такие проявления могут быть выражены очень слабо, в отдельных случаях симптомы заболевания полностью исчезают после завершения полового созревания.
Помимо кожных проявлений, синдром Блоха-Сульцбергера может приводить к развитию очаговой алопеции, дистрофии ногтей. Почти у 80% больных отмечаются аномалии зубного ряда – искривления, отсутствие зубов. В половине случаев заболевания выявляются нарушения зрения – катаракта, косоглазие, атрофия зрительного нерва и некоторые другие расстройства. Умственное развитие при синдроме Блоха-Сульцбергера обычно не страдает, но возможна некоторая задержка. В редких случаях отмечается олигофрения. Все проявления заболевания имеют тенденцию к ослаблению после завершения подросткового периода.
Диагностика и лечение синдрома Блоха-Сульцбергера
Для определения синдрома Блоха-Сульцбергера используют множество диагностических методов и техник – дерматологический осмотр, изучение наследственного анамнеза, гистологическое исследование пораженных участков кожных покровов, молекулярно-генетические анализы. При осмотре выявляются разнообразные (в зависимости от возраста больных и стадии заболевания) изменения кожи эритематозного, везикулярного или гиперкератического характера, у старших пациентов может определяться очаговая гипер- или гипопигментация кожи. Помимо этих проявлений, при синдроме Блоха-Сульцбергера возможна дистрофия ногтей, алопеция, аномалии строения зубов.
Наследственный анамнез может выявить семейный, доминантный и сцепленный с Х-хромосомой характер наследования патологии. В некоторых случаях у матери больной в анамнезе отмечается несколько случаев самопроизвольного прерывания беременности – это связано с внутриутробной смертью плода мужского пола. Результаты гистологического исследования тканей кожи при синдроме Блоха-Сульцбергера зависят от стадии заболевания – на первом этапе обнаруживается спонгиоз, развитие эпидермальных пузырей, заполненных эозинофилами и фибриновыми массами. На второй стадии патологии выявляются признаки внутриэпителиальной кератинизации, акантоз и гиперкератоз, в дерме отмечается отек с нейтрофильной и эозинофильной инфильтрацией. На третьей стадии синдрома Блоха-Сульцбергера воспалительные изменения в дерме (отек, инфильтрация) исчезают, но наблюдается значительное накопление пигмента в верхних слоях кожи. Четвертая стадия характеризуется исчезновением пигмента, развитием фиброзной ткани и частичным исчезновением придатков кожи.
Молекулярно-генетическая диагностика синдрома Блоха-Сульцбергера выполняется врачом-генетиком и может быть произведена несколькими основными техниками. Прямое автоматическое секвенирование последовательности гена IKBKG позволяет выявить практически любые изменения в его структуре. Транслокации и делеции значительных участков гена, часто выступающие в качестве причины синдрома Блоха-Сульцбергера, можно обнаружить при помощи методики FISH-анализа. Данное заболевание также может быть подтверждено посредством исследования инактивации Х-хромосом в клетках пораженных тканей.
Специфического лечения синдрома Блоха-Сульцбергера на сегодняшний момент не существует, кожные поражения на воспалительном этапе заболевания обрабатываются антисептическими средствами и растворами для предотвращения инфекционных осложнений. Кроме того, рекомендуется местное назначение глюкокортикоидных стероидов для уменьшения воспаления, однако такое лечение следует производить с осторожностью, учитывая высокую проницаемость кожных покровов у детей младшего возраста. Другие проявления синдрома Блоха-Сульцбергера (пороки развития зубов, глаз) лечат при наличии показаний.
Прогноз и профилактика
Прогноз синдрома Блоха-Сульцбергера чаще всего благоприятный, так как с момента начала полового созревания проявления заболевания значительно ослабевают. Ухудшить прогноз могут изменения внутренних органов, нервной системы, глаз, нарушения которых изредка наблюдаются при этой патологии. Кроме того, в отдельных случаях синдрома Блоха-Сульцбергера может возникать первичный иммунодефицит, который также значительно ухудшает перспективы заболевания. Профилактика патологии сводится только к пренатальной диагностике и медико-генетическому консультированию родителей при отягощенной наследственности у будущей матери.
Пигментный дерматоз (синдром Блоха-Сульцбергера) - клиника, диагностика
Пигментный дерматоз (синдром Блоха-Сульцбергера) является редким генетическим нарушением, развивающимся практически исключительно у девочек, вероятно, с доминантным путем передачи, сцепленным с Х-хромосомой, основным признаком которого является пренатальная гибель плодов мужского пола. За развитие заболевания отвечает ген NEMO — модулятор активации NF-каппа-В-пути (Fusco et al., 2004; Su et al., 2004), расположенный на Xq28 хромосоме.
а) Проявления со стороны кожи. Проявления со стороны кожи имеют необычный характер. Они представлены тремя различными, но в некоторой степени перекрывающимися стадиями (Rosman, 1992; Mangano и Barbagallo, 1993). На первой стадии изменения включают эритематозные, папулезные, везикулярные или буллезные высыпания, появляющиеся на туловище и конечностях при рождении или в течение первых двух недель жизни.
Высыпания распространяются линейно и сохраняются от дней до месяцев. На второй стадии (между второй и шестой неделями) появляются пустулезные, бородавчатые или кератозные высыпания. Они могут исчезать без следа или с образованием пигментаций, характеризующих третью стадию заболевания. Пигментные изменения могут располагаться за пределами предшествующих высыпаний, а в 10% возникают без них. Пигментация сохраняется и во взрослом возрасте, но с течением времени обычно теряет интенсивность.
При гистологическом исследовании выявляется отсутствие или сниженное содержание меланина в базальных клетках эпителия, сопровождающееся его избытком в дерме, что предполагает «утечку» или «неудержание» в базальных клетках. В 50-80% случаев имеются внекожные проявления.
б) Поражение центральной нервной системы. Поражение центральной нервной системы встречается у трети пациентов, хотя объективная частота при семейных случаях заболевания может не превышать 10% (Landy и Donnai, 1993; Hadj-Rabia et al., 2003). Припадки могут начинаться в раннем возрасте (Porksen et al., 2004) и носить генерализованный или очаговый характер с вторичной генерализацией.
В рамках ряда исследований воспалительные изменения выявлялись во всех случаях (Siemes et al., 1978, Shuper et al., 1990, Hennel et al., 2003). Возможны другие виды патологических изменений. У одного пациента было выявлено повреждение клеток передних рогов спинного мозга (Larsen et al., 1987); в другом случае отмечалось нарушение миграции (O’Doherty и Norman, 1968). Проводилась пренатальная молекулярная диагностика (Steffan et al., 2004).
в) Поражение глаз. Различные поражения глаз встречаются у трети пациентов; наиболее характерным является ретролентальная опухоль с отслойкой диспластичной сетчатки (Rosenfeld и Smith, 1983). Необходимо регулярное обследование больных детей, потому что раннее выявление и лечение ретинита может сохранить зрение (Wong et al., 2004). Аномалии зубов (зубы конической формы) встречаются у 70% пациентов.
Лечение носит симптоматический характер и включает воздействие на зрение, проивоэпилептические препараты, физиотерапию и глюкокортикоиды. Необходимо также ортопедическое лечение и специальные программы образования. Генетическое консультирование является важным компонентом лечения. Исход заболевания не всегда неблагоприятный, у некоторых пациентов отдаленные результаты лечения вполне благоприятные (Steffan et al., 2004).
Синдром Блоха-Сульцбергера отличается от сетчатого пигментного дерматоза Нигели в большей степени, чем подтипы одного заболевания (Rosman 1992). Последний встречается как среди мальчиков, так и среди девочек.
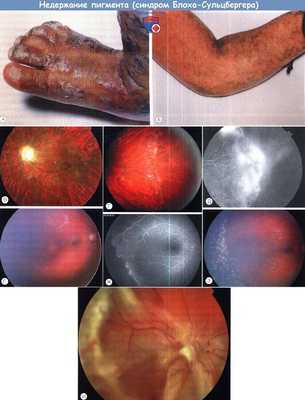
Недержание (инконтиненция) пигмента:
(А) Характерные буллезные изменения кожи с преимущественным поражением конечностей.
(Б) Буллы постепенно рассасываются, оставляя линейный участок пигменатции.
(В, Г) Характерна ретиноваскулярная патология, включающая (В) извитость сосудов сетчатки и (Г) запустевание капилляров в височной области сетчатки.
(Д) Флюоресцентная англиграфия глазного дна (ФАГ) на второй минуте видно отсутствие перфузии периферии сетчатки с неоваскуляризацией.
(E) Preretinal (цветное изображение глазного дна (Fi) и ФАГ (Fii)) и состояние после лазерноо лечения (Fiii) неперфузируемой периферии сетчатки non-perfusion.
(Ж) Отслойка сетчатки.
Болезнь Шамберга
Болезнь Шамберга (хроническая пигментная пурпура, гемосидероз кожи) — хроническое заболевание, связанное с поражением сосудистой стенки расположенных в коже капилляров и характеризующееся появлением на коже точечных кровоизлияний (петехий), переходящих в коричнево-бурые пятна. Диагностика заболевания проводится путем дерматоскопии кожи пациента. Лечение осуществляется противовоспалительными препаратами, глюкокортикоидами, при необходимости проводят гемосорбцию и плазмофорез.
Механизм возникновения болезни Шамберга до конца не ясен. Предполагают, что повреждение капилляров имеет аутоиммунную природу и связано с отложением на их стенке циркулирующих иммунных комплексов. В результате воспаления увеличивается проницаемость различных участков стенки капилляра и происходят внутрикожные петехиальные кровоизлияния. Со временем железо, содержащееся в вышедшем из сосудистого русла гемоглобине, преобразуется в пигмент гемосидерин, который и обуславливает специфический цвет пятен при болезни Шамберга.
Симптомы болезни Шамберга
Высыпания при болезни Шамберга характеризуются симметричностью и разнообразием внешнего вида. Последнее связанно с тем, что на коже одновременно с переходящими в пятна старыми элементами, появляются новые петехии. При болезнь Шамберга отсутствуют какие-либо существенные нарушения общего самочувствия. Происходит поражение только кожных капилляров, поэтому заболевание не сопровождается кровоизлияниями во внутренние органы и имеет доброкачественное течение.
Петехиальный тип болезни Шамберга проявляется появлением на кожи различного размера пятен с неровными очертаниями. Пятна состоят из мелких петехиальных кровоизлияний. Постепенно они приобретают буровато-коричневый или буровато-желтый цвет. Изменения кожи не сопровождаются отечностью и каким-либо дискомфортом. Данный тип заболевания возникает в основном у мужчин и локализуется на коже нижних конечностей.
Телеангиэктатический тип болезни Шамберга характеризуется специфическими пятнами на коже, которые напоминают медальоны. В центре таких «медальонов» расположены телеангиэктазии (сосудистые звездочки) — кистозно расширенные участки капилляров, а по периферии находятся точечные кровоизлияния.
Лихеноидный тип болезни Шамберга сочетает в себе петехиальные высыпания, коричнево-бурые пятна гемосидероза и мелкие узелки с блестящей поверхностью и практически неизмененным цветом кожи. В некоторых случаях наблюдаются только характерные узелки. Высыпания имеют множественный характер и располагаются по всему телу пациента.
Экзематоидный тип болезни Шамберга представляет собой своеобразную комбинацию высыпаний, свойственных петехиальному типу болезни Шамберга, с характерными признаками экземы. В области очагов, состоящих из точечных кровоизлияний и пятен коричнево-бурого цвета, отмечается отечность, могут появляться пузырьки, корочки и узелки. На месте высыпаний наблюдается характерный для экземы зуд.
Диагностика болезни Шамберга
Наиболее часто диагноз болезни Шамберга устанавливается на основании клинической картины и дерматоскопии области высыпаний. При исследовании сосочковой части дермы под микроскопом выявляют изменения проходящих в ней капилляров: набухание внутреннего слоя стенки капилляра, булавовидные расширения по ходу сосудов, скопления иммунных клеток и их фрагментов. О повышенной проницаемости стенки капилляра говорит выявление эритроцитов, находящихся за пределами сосудистого русла. Могут отмечаться участки изъязвления и некроза стенки капилляра, сужение его просвета из-за компенсаторного разрастания внутреннего слоя неповрежденных участков сосудистой стенки. В таких местах возникает тромбоз, значительно препятствующий кровотоку. Иногда при болезни Шамберга на стенках капилляров обнаруживаются узелки. Вместе с ними наблюдается большое количество клеток гигантских размеров.
Лечение болезни Шамберга
Пациентам с болезнью Шамберга рекомендуется соблюдать диету, исключающую аллергенные и раздражающие продукты: цитрусовые, шоколад, острые блюда, крепкий кофе и чай, жаренные и копченные блюда, спиртные напитки и т. п. Им следует избегать ушибов, переохлаждений и физических перегрузок. Большое внимание уделяется выявлению и санации очагов хронической инфекции, лечению сопутствующих заболеваний и эндокринных нарушений, поскольку они могут быть провоцирующим фактором в развитии болезни, осложнять и поддерживать ее течение.
Учитывая аутоиммунный механизм развития болезни Шамберга, в лечении применяют кортикостероиды: преднизолон, метилпреднизолон, дексаметазон и др. Используют противовоспалительные препараты, дезагреганты и ангиопротекторы. В тяжелых случаях пациентам показаны процедуры экстракорпоральной гемокоррекции: гемосорбция, плазмофорез, криопреципитация.
Синдром Блоха-Сульцбергера (Блоха—Сименса)
Синонимы: недержание пигмента, синдром Блоха—Сименса.
Определение. Заболевание Х-сцепленное доминантное, поражающее кожу, ЦНС, глаза и костную систему и характеризующееся на определенной стадии своего развития типичной меланиновой пигментацией.
Историческая справка. В 1906 г. А. Е. Garrod (1857—1936) написал отчет об умственно отсталой девочке с тетраплегией и характерной пигментацией кожи. В 1926 г. швейцарский дерматолог Бруно Блох (1878—1933) описал случай данного заболевания и первым предложил термин «недержание пигмента». Более детально изучил заболевание американский дерматолог профессор Марион Сульцбергер (1895-1983) в 1927 г. Он описал своеобразные эктодермальные и мезодермальные дефекты, которые возникают в младенческом возрасте и проявляются буллезной сыпью, а в дальнейшем характерной пигментацией кожи. В 1929 г. появилась еще одна публикация, которую выполнил H.W.Siemens. В 1989 г. A. Sefiani с соавторами выявили локализацию гена в хромосоме Xq28 при данном синдроме. В 1993 г. была выполнена публикация S.J. Landy и D. Donnai с обширным обзором случаев с синдромом Блоха—Сульцбергера.
Этиология и патогенез. Генетическое заболевание наследуется доминантно и сцеплено с полом. Характеризуется нарушением меланогенеза. Развитие пигментации связано с тем, что имеется функциональная аномалия пигментообразующих клеток базального слоя эпидермиса. Из-за патологической проницаемости клеточной мембраны для меланина эти клетки отдают в дерму весь или почти весь вырабатываемый ими меланин. Линейные образования на коже при данном синдроме обусловлены мозаицизмом вследствие инактивации Х-хромосомы. Были выявлены два генетических локуса при этом синдроме. При I типе ген локализуется в хромосоме Xq11, болезнь характеризуется спорадическими случаями, генетические аномалии возникают без предшествующей воспалительной фазы. При II типе (семейный вариант заболевания) ген расположен в хромосоме Xq28. При этом типе синдрома отмечаются Х-сцепленные доминантные мутации в гене NEMO.
Предполагают, что эти мутации приводят к снижению иммунной толерантности в эктодермальных тканях. У гетерозиготных девочек это вызывает реакцию, похожую на аутоиммунную, а у гомозиготных мальчиков — реакцию по типу «трансплантат против хозяина», приводящую к фатальному исходу заболевания. Развитию заболевания способствуют внутриутробные травматизация, аллергизация и инфекция плода, нейрогуморальные нарушения. Возраст и пол. В 95—97% случаев регистрируется только у лиц женского пола. Плод мужского пола обычно погибает внутриутробно. В мировой литературе отмечено выживание мальчиков в 30 случаях. Дети уже рождаются с клиническими проявлениями или заболевание начинается в течение первых 6 недель жизни. Поражения кожи. В течении синдрома выделяют четыре стадии. Первая является воспалительной (везикулярной).
Длится от момента рождения и в течение нескольких первых месяцев жизни. Имеются эритематозные, уртикарные, пузырьковые и пузырные элементы на коже туловища и (преимущественно) конечностей. Лицо, ладони, подошвы и слизистые не поражаются.
Высыпания имеют тенденцию к полосовидному, вихревидному расположению. Вторая стадия называется веррукозной и длится до первого года жизни. На тех же участках появляются лихеноидные, лентикулярные папулы и веррукозные высыпания. На 3—6-м месяце жизни папулы постепенно рассасываются. Вторая стадия отмечается у 2/3 больных. Для третьей стадии характерны гиперпигментации.
Длится с одного года до подросткового возраста. Эта стадия присутствует у всех пациентов с синдромом Блоха-Сульцбергера. На фоне папулезной сыпи или в период ее разрешения появляются пигментные пятна от темно-серого до темно-коричневого цвета, иногда с ливедо-синеватым оттенком. Гиперпигментации имеют полосовидную, сетчатую, паукообразную форму или в виде завихрений. Очаги располагаются по линиям Блашко (но не по дерматомам или линиям расщепления кожи). Гиперпигментации локализуются на голове, туловище и конечностях, часто напоминают брызги. Примерно у 40% больных возникновение пигментации не связано с предшествующими высыпаниями. Для четвертой стадии характерны гипопигментации. Между периодом полового созревания и 25—30 годами гиперпигментации спонтанно исчезают, оставляя легкую атрофию с гипопигментацией. В этих зонах отсутствуют волосы и потовые железы. Такие изменения отмечают преимущественно на нижних конечностях.
Другие изменения кожи и ее придатков. В части случаев у больных обнаруживают очаги рубцовой алопеции на волосистой части головы, иногда дистрофию ногтей (около 10% случаев), диффузную алопецию.
Офтальмологические изменения. Микрофтальмия, аномалия сосудов сетчатки, псевдоглиома, катаракта, атрофия зрительного нерва, глубокий или поверхностный кератит, отслойка сетчатки, косоглазие.
Изменения опорно-двигательной системы. Аномалии черепа, сколиоз, врожденный вывих бедра, асимметрия грудной клетки.
Неврологические изменения. Замедление мышления (16%), эпилепсия (3%), спастические параличи (13%), гидро- и/или микроцефалия, олигофрения.
Другие изменения. Легочная гипертензия, добавочные соски, врожденные пороки сердца, отсутствие или поражение зубов, преимущественно премоляров и верхних боковых резцов (замедление прорезывания, патология дентина, коническая форма). Во время воспалительной фазы в крови и пузырной жидкости выявляют эозинофилию (50—74%), лейкоцитоз периферической крови.
Гистология. В I стадии наблюдают спонгиоз с образованием пузырьков и пузырей, содержащих эозинофилы, фибрин. Местами рассеяны крупные дискератотические клетки с гомогенной протоплазмой. Дерма инфильтрирована мононуклеарами и эозинофилами.
Во II стадии картина может быть сходна с псевдоэпителиоматозной гиперплазией. Отмечаются акантоз, папилломатоз и гиперкератоз с внутриэпидермальной кератинизацией (кератиноциты располагаются спирально), местами — вакуольная дистрофия базальных клеток. В дерме — отек и инфильтраты из гистиоцитов, лимфоцитов, нейтрофильных и эозинофильных гранулоцитов, много меланофоров. В III стадии воспалительные явления уменьшаются или исчезают. В верхних частях собственно кожи, главным образом в сосочковом слое, отмечается обильное скопление пигмента. Заполненные пигментом соединительнотканные клетки так густо лежат на самой дермоэпидермальной границе, что получается впечатление их «контакта» с базальными клетками. В клетках базального слоя меланина мало или содержание его в пределах нормы, в них отмечается вакуолизация. Выявляются зоны истончения эпидермиса, очаговый гиперкератоз. В IV стадии — снижение количества меланина, фиброзные изменения и отсутствие или снижение придатков кожи в пораженных областях.
Диагноз ставят по клинической картине кожных поражений в зависимости от стадии заболевания, а также на основании результатов инструментальных и генетических исследований.
Дифференцируют на воспалительной стадии с герпесом, буллезными дерматозами: эпидермолизом, линейным IgA и пемфигоидом. На веррукозной стадии дифференцируют с эпидермальным невусом, на стадии гиперпигментации — с линейным и завитым невоидным гипермеланозом, на стадии гипопигментации — с гипомеланозом Ито.
Течение и прогноз. У 60—80% пациентов с этим синдромом имеются системные аномалии экто- и мезодермального происхождения. При возникновении припадков на первой неделе жизни прогноз сомнительный, отмечается задержка развития. Отсутствие припадков и соответствие развития возрастным нормам свидетельствуют о благоприятном прогнозе. Пигментации персистируют на протяжении многих лет и постепенно исчезают в подростковом или раннем взрослом возрасте.
Лечение. В воспалительной стадии в виде наружного лечения используют глюкокортикоиды. При образовании пузырьков накладывают повязки, смоченные антисептическим раствором (бледно-розовый раствор калия перманганата). При выраженных воспалительных изменениях назначают глюкокортикоиды внутрь, например преднизолон 0,5—1,0 мг/кг в день с быстрой отменой. Проводят профилактику вторичной инфекции. Поражения других органов лечат у смежных специалистов.
Читайте также: