Почечный канальцевый ацидоз
Добавил пользователь Skiper Обновлено: 29.01.2026
Похожие темы научных работ по клинической медицине , автор научной работы — Аксенова М.Е., Кузнецова С.А., Хабарадзе М.Н., Николаева Е.А.
Факторы риска поражения сердечно-сосудистой системы у детей с аутосомно-доминантной поликистозной болезнью почек
Влияние массы тела и артериальной гипертензии на структурную перестройку сердца при хроническом гломерулонефрите
Особенности структурно-функциональных нарушений левого желудочка у больных подагрой в сочетании с артериальной гипертензией и хронической болезнью почек
Текст научной работы на тему «Почечный тубулярный ацидоз, 3 тип»
Раздел 7. Нефрология
ПОЧЕЧНЫЙ ТУБУЛЯРНЫЙ АЦИДОЗ, 3 ТИП
Аксенова М.Е., Кузнецова С.А., Хабарадзе М.Н., Николаева Е.А.
НИКИ педиатрии им. академика Ю.Е. Вельтищева ФГБОУ ВО РНИМУ имени Н.И. Пирогова Минздрава России, г. Москва
Клиническое наблюдение. Родные сибсы 12 и 13 лет от близкородственного брака, с 6 мес — задержка физического развития, с 1 года — отставание в психомоторном развитии. На момент обследования: рост 6, осадок — норма, экскреция кальция, магния, оксалатов, уратов, фосфора в норме; УЗИ почек: незначительное нарушение диффе-ренцировки паренхимы; рентген трубчатых костей и черепа: остеопороз, признаки рахитоподобного заболевания, остеопетроз; МРТ головного мозга: множественные кальцинаты с поражением базальных ядер, хвостатого тела, таламуса; результаты молеку-лярно-генетического исследования: мутация гена CA2 (c.82C>T). Мальчикам была назначена терапия бикарбонатом натрия в сочетании с цитратом калия.
Заключение: Пациентам с почечным тубулярным ацидозом для уточнения диагноза и определения тактики ведения целесообразно проводить рентгенологическое и неврологическое обследование.
ОЖИРЕНИЕ КАК ФАКТОР РИСКА КАРДИОВАС-КУЛЯРНЫХ ОСЛОЖНЕНИЙ У ДЕТЕЙ С ХРОНИЧЕСКИМИ БОЛЕЗНЯМИ ПОЧЕК
Аксенова М.Е., Тутельман К.М., Конькова Н.Е., Лепаева Т.В., Кузнецова Т.А.
НИКИ педиатрии им. академика Ю.Е. Вельтищева
ФГБОУ ВО РНИМУ имени Н.И. Пирогова Минздрава России, г. Москва
Известно, что ожирение является модифицируемым фактором риска развития патологии почек и кардиоваскулярных заболеваний. Логично ожидать, что ожирение через активацию ренин-альдо-стероновой системы, оксидативного стресса, дисли-пидемию и асептическое воспаление также влияет на развитие кардиоваскулярных осложнений у детей с хроническими болезнями почек (ХБП).
Цель. Определить значение ожирения для формирования кардиоваскулярной патологии у детей с ХБП.
Материалы и методы. В исследование вошло 355 детей 3—17 лет с ХБП. Всем детям проводилось кли-нико-лабораторное обследование, включая определение индекса массы тела (ИМТ), уровня разового артериального давления (АД) методом Короткова, расчет скорости клубочковой фильтрации (рСКФ) по формуле Шварца, проведение суточного мониторирования артериального давления (СМАД) на аппаратах ABPM («Медитек», Венгрия) и BPLab («Петр Телегин», Россия). ЭХО-КГ с определением массы миокарда левого желудочка (ММЛЖ) по формуле Devereux RB, индекса массы миокарда левого желудочка (ИММЛЖ) по формуле De Simone G, относительной толщины стенки левого желудочка (ОТСЛЖ) была проведена 180 детям. За нормальные значения ИМТ, АД, ММЛЖ, ИММЛЖ, ОТСЛЖ приняты значения 95% (М/Д=0,76; возраст 13±1,95 лет; рСКФ=105±30,8 мл/мин/1,73 м2).
Заключение. Получено, что ожирение повышает риск развития артериальной гипертензии и гипертрофии миокарда левого желудочка у детей с хроническими болезнями почек.
ФУНКЦИЯ ПОЧЕК У ДЕТЕЙ ПРИ ПОЛИКИСТО-ЗЕ С АУТОСОМНО-ДОМИНАНТНЫМ ТИПОМ НАСЛЕДОВАНИЯ
Андреева Э.Ф., Денисова И.В.
ФГБОУ ВО СПбГПМУ Минздрава России, г. Санкт-Петербург
Цель: оценить функцию почек у детей при поли-кистозе почек с аутосомно-доминантным типом на-
РОССИЙСКИЙ ВЕСТНИК ПЕРИНАТОЛОГИИ И ПЕДИАТРИИ, 2017; 62:(4) ROSSIYSKIY VESTNIK PERINATOLOGY IPEDIATRII, 2017; 62:(4)
Публикации в СМИ
Почечный канальцевый ацидоз (ПКА) — синдром, характеризующийся метаболическим ацидозом, развивающимся вследствие нарушения подкисления мочи почками в отсутствие выраженного снижения функции клубочкового аппарата (нарушение секреции ионов водорода в дистальных или реабсорбции бикарбонатов в проксимальных отделах канальцев почек, что приводит к хроническому метаболическому ацидозу, гипокалиемии, нефрокальцинозу и развитию рахита или остеомаляции).
Классификация. Выделяют два типа ПКА • Проксимальный ПКА формируется вследствие снижения способности проксимальных почечных канальцев реабсорбировать профильтровавшиеся бикарбонаты • Дистальный ПКА •• Гипокалиемический (классический) — развивается вследствие нарушения секреции водородных ионов дистальными канальцами •• Гиперкалиемический — развивается вследствие первичного или вторичного дефицита альдостерона или резистентности к нему.
Статистические данные. Точных данных о распространённости нет. Проксимальный и дистальный гипокалиемический ПКА возникают преимущественно у детей, гиперкалиемический ПКА — у взрослых.
Этиология. ПКА может быть наследственным, развиваться при токсическом, лекарственном повреждении канальцев, при общем или системном заболевании • Проксимальный ПКА •• Лекарственный (тетрациклин, ацетазоламид, сульфаниламиды, соли тяжёлых металлов) •• Наследственный (при синдроме Фанкони, болезни Уилсона, синдроме Лоу) •• Амилоидоз •• Множественная миелома •• Дефицит витамина D • Дистальный классический ПКА •• Лекарственный (амфотерицин B, препараты лития, НПВС) •• Наследственный •• Состояние после трансплантации почки •• Синдром Шёгрена •• Криоглобулинемия •• Фиброзирующий альвеолит •• Хронический активный гепатит •• СКВ •• Первичный билиарный цирроз печени •• Первичный гиперпаратиреоз •• Отравление витамином D •• Гипертиреоз •• Медуллярная губчатая почка •• Пиелонефрит •• Обструкция мочевых путей • Гиперкалиемический ПКА •• Волчаночная нефропатия •• Диабетическая нефропатия •• Гипоальдостеронизм •• Нефросклероз вследствие артериальной гипертензии •• Тубуло-интерстициальные нефропатии •• Болезнь Аддисона •• Острая надпочечниковая недостаточность.
Генетические особенности • ПКА с прогрессирующей нейросенсорной тугоухостью (267300, ATP6B1, VPP3, r ) • Дистальный ПКА (179800, SLC4A1, AE1, EPB3, Â , также r ) • Дистальный ПКА, аутосомно-рецессивный, без нарушения слуха (602722, ATP6V0A4, ATP6N1B, VPP2, RTA1C, RTADR, r ) • Проксимальный ПКА с глазными аномалиями (604278, SLC4A4, NBC1, KNBC, SLC4A5, r ) • Синдром остеопетроза с ПКА (*259730, CA2, r ).
Патогенез • Проксимальный ПКА формируется при выраженном снижении способности проксимальных канальцев реабсорбировать профильтровавшиеся бикарбонаты; ацидоз редко бывает изолированным, чаще сочетается с другими канальцевыми дисфункциями. Характерна гипокалиемия вследствие гиперальдостеронизма • При дистальном ПКА нарушена секреция водородных ионов дистальными канальцами: при гипокалиемическом синтез альдостерона сохранён, при гиперкалиемическом — нарушен (либо повышена резистентность канальцев к альдостерону).
Патоморфология. При изолированном ПКА изменения могут отсутствовать. При вторичном ПКА или синдроме Фанкони — дистрофия и атрофия канальцевого эпителия.
Клинические проявления • При изолированном проксимальном ПКА клинические проявления могут отсутствовать; у детей возможны задержка роста, гипорефлексия, мышечная слабость; изменения обмена кальция минимальны и компенсированы • При первичном (наследственном) дистальном ПКА первые признаки болезни могут появиться в возрасте 2–3 лет •• Отставание в росте •• Мышечная слабость, рвота, снижение рефлексов, вплоть до параличей, связаны с хроническим метаболическим ацидозом и гипокалиемией •• Одновременное увеличение мобилизации кальция из кости и гиперкальциурия приводят к остеопении, болям в костях, затруднениям при ходьбе •• Формирующийся нефрокальциноз сопровождается развитием пиелонефрита и формированием ХПН. Для развёрнутой картины заболевания характерны полиурия и гипоизостенурия • При синдроме Фанкони •• У детей ••• Полиурия, жажда, дегидратация, иногда судороги (гипокальциемия) ••• Мышечная слабость (потеря калия) ••• Отставание в физическом, иногда в умственном развитии; рахит ••• Инфекции •• При начале болезни во взрослом возрасте ••• Полиурия ••• Мышечная слабость ••• Боли в костях, переломы ••• При прогрессировании — артериальная гипертензия, ХПН.
Сопутствующая патология • Дистальный ПКА у детей: рахит, нефрокальциноз и нефролитиаз • Дистальный ПКА у взрослых: аутоиммунные заболевания (например, синдром Шёгрена) • Проксимальный ПКА: синдром Фанкони.
Лабораторные исследования • Снижение рН крови: метаболический ацидоз • Повышение рН мочи • Снижение концентрации бикарбонатов в крови • Бикарбонатурия при проксимальном ПКА • Гипокалиемия при проксимальном и дистальном классическом ПКА, гиперкалиемия при дистальном (реже) • Гипохлоремия при дистальном и гиперхлоремия при проксимальном ПКА • Гипокальциемия, гиперкальциурия при дистальном ПКА • Рахитоподобные изменения в костях из-за потери кальция при дистальном ПКА • Остеомаляция из-за системного ацидоза • Уровни ренина и альдостерона при проксимальном ПКА повышены, при дистальном — увеличены или снижены • Азот мочевины крови и креатинин: для исключения почечной недостаточности • Бактериологическое исследование мочи: для исключения инфицирования мочевыводящих путей микробами, расщепляющими мочевину.
Специальные исследования необходимы для исключения потенциальных осложнений • Внутривенная пиелография: нефрокальциноз, нефролитиаз, врождённые заболевания почек • УЗИ для оценки размеров почек или выявления камней • Рентгенография костей: остеомаляция.
Особенности течения у детей. Возможны задержка роста, отставание в умственном развитии при синдроме Фанкони.
Диагностическая тактика • Диагноз проксимального ПКА ставят на основании выявления гиперхлоремического ацидоза, бикарбонатурии, щелочной реакции мочи; при нагрузке хлоридом аммония рН мочи становится ниже 6,0, т.к. сохраняется способность дистальных канальцев к подкислению мочи • Диагноз проксимального ПКА ставят на основании выявления метаболического ацидоза, щелочной реакции мочи, однако при нагрузке хлоридом аммония или хлоридом кальция рН мочи не становится ниже 6,0.
Дифференциальная диагностика • Метаболический ацидоз иной этиологии •• Диарея с потерей бикарбоната с калом •• Ацидоз при ХПН •• Респираторный ацидоз • Мочекаменная болезнь • Инфицирование мочевых путей микробами- щелочеобразователями (протей и др.).
ЛЕЧЕНИЕ
Тактика ведения • Адекватная медикаментозная коррекция ацидоза • Ограничение поваренной соли •• У детей ограничение поступления поваренной соли необходимо в любом случае • Изолированный ПКА у взрослых не требует ограничения соли.
Лекарственная терапия • Проксимальный ПКА •• Натрия гидрокарбонат до 1 г/кг/сут (до 10 ммоль/кг/сут) внутрь в 2–3 приёма •• Калия бикарбонат при гипокалиемии •• Цитратные смеси (поглощают Н + ) •• Тиазидные диуретики (гидрохлоротиазид 25–50 мг/сут) повышают реабсорбцию бикарбонатов • Дистальный ПКА •• Натрия гидрокарбонат в дозе 0,2 г/кг/сут (1–3 ммоль/кг/сут) в 4–6 приёмов •• При гипокалиемии препараты калия, при гиперкалиемии фуросемид •• Дезоксикортон (2,5–5 мг через день) — при недостаточности минералокортикоидов или резистентности к ним.
Профилактика вторичного ПКА — лечение основного заболевания
Течение и прогноз • При изолированном дефекте прогноз благоприятный • При наследственных синдромах и возникновении заболевания в детском возрасте — прогноз серьёзный • При вторичном ПКА прогноз зависит от основного заболевания • При вторичном синдроме Фанкони и дистальном ПКА возможно прогрессирование заболевания почек с развитием артериальной гипертензии и ХПН.
Возможные осложнения • Нефрокальциноз • Пиелонефрит • Нефролитиаз • Рахит у детей, остеомаляция у взрослых • ХПН.
Сокращение. ПКА — почечный канальцевый ацидоз.
МКБ-10. N25.8 Другие нарушения, обусловленные дисфункцией почечных канальцев
Код вставки на сайт
Ацидоз почечный канальцевый
Почечный канальцевый ацидоз (ПКА) — синдром, характеризующийся метаболическим ацидозом, развивающимся вследствие нарушения подкисления мочи почками в отсутствие выраженного снижения функции клубочкового аппарата (нарушение секреции ионов водорода в дистальных или реабсорбции бикарбонатов в проксимальных отделах канальцев почек, что приводит к хроническому метаболическому ацидозу, гипокалиемии, нефрокальцинозу и развитию рахита или остеомаляции).
Классификация. Выделяют два типа ПКА • Проксимальный ПКА формируется вследствие снижения способности проксимальных почечных канальцев реабсорбировать профильтровавшиеся бикарбонаты • Дистальный ПКА •• Гипокалиемический (классический) — развивается вследствие нарушения секреции водородных ионов дистальными канальцами •• Гиперкалиемический — развивается вследствие первичного или вторичного дефицита альдостерона или резистентности к нему.
Статистические данные. Точных данных о распространённости нет. Проксимальный и дистальный гипокалиемический ПКА возникают преимущественно у детей, гиперкалиемический ПКА — у взрослых.
Этиология. ПКА может быть наследственным, развиваться при токсическом, лекарственном повреждении канальцев, при общем или системном заболевании • Проксимальный ПКА •• Лекарственный (тетрациклин, ацетазоламид, сульфаниламиды, соли тяжёлых металлов) •• Наследственный (при синдроме Фанкони, болезни Уилсона, синдроме Лоу) •• Амилоидоз •• Множественная миелома •• Дефицит витамина D • Дистальный классический ПКА •• Лекарственный (амфотерицин B, препараты лития, НПВС) •• Наследственный •• Состояние после трансплантации почки •• Синдром Шёгрена •• Криоглобулинемия •• Фиброзирующий альвеолит •• Хронический активный гепатит •• СКВ •• Первичный билиарный цирроз печени •• Первичный гиперпаратиреоз •• Отравление витамином D •• Гипертиреоз •• Медуллярная губчатая почка •• Пиелонефрит •• Обструкция мочевых путей • Гиперкалиемический ПКА •• Волчаночная нефропатия •• Диабетическая нефропатия •• Гипоальдостеронизм •• Нефросклероз вследствие артериальной гипертензии •• Тубуло-интерстициальные нефропатии •• Болезнь Аддисона •• Острая надпочечниковая недостаточность.
Генетические особенности • ПКА с прогрессирующей нейросенсорной тугоухостью (267300, ATP6B1, VPP3, r ) • Дистальный ПКА (179800, SLC4A1, AE1, EPB3, Â , также r ) • Дистальный ПКА, аутосомно-рецессивный, без нарушения слуха (602722, ATP6V0A4, ATP6N1B, VPP2, RTA1C, RTADR, r ) • Проксимальный ПКА с глазными аномалиями (604278, SLC4A4, NBC1, KNBC, SLC4A5, r ) • Синдром остеопетроза с ПКА (*259730, CA2, r ).
Патогенез • Проксимальный ПКА формируется при выраженном снижении способности проксимальных канальцев реабсорбировать профильтровавшиеся бикарбонаты; ацидоз редко бывает изолированным, чаще сочетается с другими канальцевыми дисфункциями. Характерна гипокалиемия вследствие гиперальдостеронизма • При дистальном ПКА нарушена секреция водородных ионов дистальными канальцами: при гипокалиемическом синтез альдостерона сохранён, при гиперкалиемическом — нарушен (либо повышена резистентность канальцев к альдостерону).
Патоморфология. При изолированном ПКА изменения могут отсутствовать. При вторичном ПКА или синдроме Фанкони — дистрофия и атрофия канальцевого эпителия.
Клинические проявления • При изолированном проксимальном ПКА клинические проявления могут отсутствовать; у детей возможны задержка роста, гипорефлексия, мышечная слабость; изменения обмена кальция минимальны и компенсированы • При первичном (наследственном) дистальном ПКА первые признаки болезни могут появиться в возрасте 2–3 лет •• Отставание в росте •• Мышечная слабость, рвота, снижение рефлексов, вплоть до параличей, связаны с хроническим метаболическим ацидозом и гипокалиемией •• Одновременное увеличение мобилизации кальция из кости и гиперкальциурия приводят к остеопении, болям в костях, затруднениям при ходьбе •• Формирующийся нефрокальциноз сопровождается развитием пиелонефрита и формированием ХПН. Для развёрнутой картины заболевания характерны полиурия и гипоизостенурия • При синдроме Фанкони •• У детей ••• Полиурия, жажда, дегидратация, иногда судороги (гипокальциемия) ••• Мышечная слабость (потеря калия) ••• Отставание в физическом, иногда в умственном развитии; рахит ••• Инфекции •• При начале болезни во взрослом возрасте ••• Полиурия ••• Мышечная слабость ••• Боли в костях, переломы ••• При прогрессировании — артериальная гипертензия, ХПН.
Сопутствующая патология • Дистальный ПКА у детей: рахит, нефрокальциноз и нефролитиаз • Дистальный ПКА у взрослых: аутоиммунные заболевания (например, синдром Шёгрена) • Проксимальный ПКА: синдром Фанкони.
Лабораторные исследования • Снижение рН крови: метаболический ацидоз • Повышение рН мочи • Снижение концентрации бикарбонатов в крови • Бикарбонатурия при проксимальном ПКА • Гипокалиемия при проксимальном и дистальном классическом ПКА, гиперкалиемия при дистальном (реже) • Гипохлоремия при дистальном и гиперхлоремия при проксимальном ПКА • Гипокальциемия, гиперкальциурия при дистальном ПКА • Рахитоподобные изменения в костях из-за потери кальция при дистальном ПКА • Остеомаляция из-за системного ацидоза • Уровни ренина и альдостерона при проксимальном ПКА повышены, при дистальном — увеличены или снижены • Азот мочевины крови и креатинин: для исключения почечной недостаточности • Бактериологическое исследование мочи: для исключения инфицирования мочевыводящих путей микробами, расщепляющими мочевину.
Специальные исследования необходимы для исключения потенциальных осложнений • Внутривенная пиелография: нефрокальциноз, нефролитиаз, врождённые заболевания почек • УЗИ для оценки размеров почек или выявления камней • Рентгенография костей: остеомаляция.
Особенности течения у детей. Возможны задержка роста, отставание в умственном развитии при синдроме Фанкони.
Диагностическая тактика • Диагноз проксимального ПКА ставят на основании выявления гиперхлоремического ацидоза, бикарбонатурии, щелочной реакции мочи; при нагрузке хлоридом аммония рН мочи становится ниже 6,0, т.к. сохраняется способность дистальных канальцев к подкислению мочи • Диагноз проксимального ПКА ставят на основании выявления метаболического ацидоза, щелочной реакции мочи, однако при нагрузке хлоридом аммония или хлоридом кальция рН мочи не становится ниже 6,0.
Дифференциальная диагностика • Метаболический ацидоз иной этиологии •• Диарея с потерей бикарбоната с калом •• Ацидоз при ХПН •• Респираторный ацидоз • Мочекаменная болезнь • Инфицирование мочевых путей микробами- щелочеобразователями (протей и др.).
ЛЕЧЕНИЕ
Тактика ведения • Адекватная медикаментозная коррекция ацидоза • Ограничение поваренной соли •• У детей ограничение поступления поваренной соли необходимо в любом случае • Изолированный ПКА у взрослых не требует ограничения соли.
Лекарственная терапия • Проксимальный ПКА •• Натрия гидрокарбонат до 1 г/кг/сут (до 10 ммоль/кг/сут) внутрь в 2–3 приёма •• Калия бикарбонат при гипокалиемии •• Цитратные смеси (поглощают Н + ) •• Тиазидные диуретики (гидрохлоротиазид 25–50 мг/сут) повышают реабсорбцию бикарбонатов • Дистальный ПКА •• Натрия гидрокарбонат в дозе 0,2 г/кг/сут (1–3 ммоль/кг/сут) в 4–6 приёмов •• При гипокалиемии препараты калия, при гиперкалиемии фуросемид •• Дезоксикортон (2,5–5 мг через день) — при недостаточности минералокортикоидов или резистентности к ним.
Профилактика вторичного ПКА — лечение основного заболевания
Течение и прогноз • При изолированном дефекте прогноз благоприятный • При наследственных синдромах и возникновении заболевания в детском возрасте — прогноз серьёзный • При вторичном ПКА прогноз зависит от основного заболевания • При вторичном синдроме Фанкони и дистальном ПКА возможно прогрессирование заболевания почек с развитием артериальной гипертензии и ХПН.
Возможные осложнения • Нефрокальциноз • Пиелонефрит • Нефролитиаз • Рахит у детей, остеомаляция у взрослых • ХПН.
Сокращение. ПКА — почечный канальцевый ацидоз.
МКБ-10. N25.8 Другие нарушения, обусловленные дисфункцией почечных канальцев
Почечный канальцевый ацидоз
Определение. ПКА — многочисленная группа заболеваний, в основе которых лежит неспособность почечных канальцев подкислять мочу и сопровождающихся сдвигом реакции крови в кислую сторону. В конечном итоге патология характеризуется метаболическим ацидозом, полиурией, рахитоподобными изменениями, нефрокальцинозом и нефролитиазом.
Синонимы названия ПКА — синдром Лайтвуда–Олбрайта, гиперхлоремический тубулярный ацидоз с гиперкальциурией и гипоцитратурией.
Семейный анамнез. Типы наследственной передачи различные. Это генетически гетерогенные заболевания.
Патогенез. В настоящее время известно, что механизмы развития ПКА связаны, главным образом, с двумя процессами: избыточной потерей бикарбонатов с мочой или недостаточной секрецией водородных ионов в просвет почечных канальцев.
Классификация. В основу положены этиопатогенетические, клини- ко-функциональные принципы.
ПКА у детей принято классифицировать:
– на первичный (наследственный);
– вторичный (обусловленный рядом заболеваний). По локализации дефекта ПКА бывает:
– дистальный (I тип, первичный и вторичный);
– проксимальный (II тип, первичный и вторичный);
– проксимальный и дистальный (III тип);
– ПКА с гиперкалиемией (IV тип).
– отставание в физическом развитии;
– выраженный метаболический ацидоз;
– рахитоподобные изменения костей (чаще в виде вальгусной деформации нижних конечностей);
– изменение ионограммы крови (гипокалиемия, гипокальциемия, гипонатриемия, гиперхлоремия, умеренная гипофосфатемия);
– низкие величины экскреции ионов водорода с мочой;
– нефрокальциноз и нефролитиаз.
– длительный стойкий субфебрилитет;
ПКА I типа (дистальный, классический). При этом варианте
дистальный каналец не способен создавать определенный градиент концентрации водородных ионов между кровью и канальцевой жидкостью, нарушена их секреция в дистальных канальцах и снижено содержание. Резко нарушается аммоний и титруемая кислотность мочи. рН мочи обычно не бывает менее 6,8 независимо от тяжести ацидоза. Особенностью этой формы является то, что в мочу постоянно выделяются гидрокарбонаты, вместо которых в кровь диффундируют ионы хлора — развивается гиперхлоремия.
Для классического ПКА I типа характерно:
– отставание в росте;
– рахитоподобные изменения в костях;
– метаболический ацидоз (дефицит ВЕ равен от –10 до 20 ммоль/л);
– кризы обезвоживания и полиурия;
– нефрокальциноз (двусторонний) и мочекаменная болезнь с тубулоинтерстициальным нефритом и/или пиелонефритом;
– повышение уровня хлоридов крови;
– щелочная реакция мочи (рН = 6,7–7,2);
– гиперкальциурия до 10–20 мг/сут (при норме 1–5 мг/сут). Полиурия при ПКА I типа, обусловленная повышенной экскрецией
натрия, приводит к гиперкалиурии, гиперкальциурии. Большая потеря кальция с мочой и ее щелочная реакция приводит к отложению камней в почках, к 3–5 годам возникает двусторонний нефрокальциноз, который создает условия для вторичной инфекции мочевыводящей системы или абактериального процесса.
Гипокальциемия стимулирует выработку ПТГ, развивается вторичный гиперпаратиреоз, что приводит к резорбции костной ткани. В результате этого в кровь поступает кальций, поэтому отмечается нормокальциемия. Развиваются деформации (Х-образные искривления нижних конечностей, бочкообразная форма грудной клетки и др.).
I тип ПКА встречается одинаково как у мальчиков, так и у девочек. Чаще впервые проявляется в 2–3 года жизни. Однако этот тип может встречаться и у взрослых (так называемая «форма взрослых»). Микросимптомы могут проявляться значительно раньше — до манифестации этого заболевания, в грудном возрасте. К ним можно отнести:
– недостаточную прибавку веса;
– признаки дегидратации на фоне полиурии и полидипсии;
– беспричинный длительный субфебрилитет. Деформации костной системы развиваются к 2–3 годам.
Для «неполных» форм I типа ПКА характерен нефрокальциноз.
Системного ацидоза нет. Отмечается нарушение к подкислению мочи, но нет резкого снижения секреции аммония, что компенсирует ограниченную экскрецию титруемых кислот.
ПКА I типа делят на 3 подтипа:
1. ПКА с аутосомно-доминантной наследственной передачей .
Клинические проявления (в конце первого года жизни ребенка): рвота (периодически), гипотрофия, запоры, жажда, полиурия, немотивированные повышения температуры, задержка статико-моторного развития,
позднее прорезывание зубов. Через 1,5–2 года присоединяются рахитоподобные изменения скелета (преимущественно в виде вальгусных деформаций нижних конечностей), деформации грудной клетки, рахитических «четок», «браслеток». У подавляющего большинства пациентов с данным типом ПКА развивается двусторонний нефрокальциноз.
2. ПКА с прогрессирующей глухотой . Это аутосомно-рецессивный тип наследования. Довольно часто при нем наблюдается кровное родство родителей.
В клинической картине отмечаются признаки, характерные для ПКА дистального типа, а также нейросенсорная глухота различной степени тяжести (снижение слуха от 100 до 50 дБ), что может быть причиной задержки темпов психомоторного развития.
В крови при метаболическом ацидозе различной степени выраженности наблюдается нормальный уровень кальция. Реакция мочи щелочная.
3. ПКА без глухоты . Тип наследования — аутосомно-рецессивный. Клинические проявления те же, что при аутосомно-рецессивном типе
с глухотой (гиперхлоремический метаболический ацидоз, двусторонний нефрокальциноз/уролитиаз, щелочная реакция мочи, бикарбонатурия, гиперкальциурия, гипоцитратурия, снижение мочевой экскреции аммония).
ПКА II типа (проксимальный канальцевый ацидоз). В его основе лежит повышенная экскреция бикарбонатов с мочой и нарушение их реабсорбции в проксимальных канальцах, причины которого различны (недостаточность карбоангидразы II-(С); снижение активности в крови карбоангидразы I-(В); дисплазия почек и др.).
За сутки с мочой у больных выделяется до 10–15 % бикарбонатов клубочкового фильтрата, в то время как у здоровых — до 2 %. В связи
с выраженной потерей бикарбонатов содержание буферных оснований в крови значительно снижается. Секреция Н + при этом типе ПКА не нарушена, поэтому реакция мочи может быть кислой, экскреция аммония
с мочой и ее титруемая кислотность без отклонений.
Клинически это заболевание проявляется в 3–18 мес. с таких симптомов, как:
Почечный тубулярный ацидоз ( Ацидоз почечных канальцев , Почечный канальцевый ацидоз )
Почечный тубулярный ацидоз – это разновидность тубулопатии, которая сопровождается дисбалансом электролитов из-за сбоя экскреции ионов водорода (I), нарушением обратного всасывания бикарбонатов (II), аномальной продукцией альдостерона или взаимодействия с ним (IV). Тип III встречается казуистически редко. Течение может быть бессимптомным или с проявлениями электролитных расстройств: слабостью, тошнотой, костными деформациями. Диагностика почечной ацидемии базируется на измерениях рН мочи и электролитов, данных генетических анализов, УЗИ. Лечение предполагает восстановление кислотно-щелочного баланса: ощелачивающие средства, устранение гипо- гиперкалиемии и пр.
МКБ-10
N25.8 Другие нарушения, обусловленные дисфункцией почечных канальцев. Почечно-канальцевый ацидоз БДУ.
Общие сведения
Почечный канальцевый ацидоз (RTA, тубулярный, трубчатый, ацидоз почечных канальцев) определяется нефрологами как синдром, при котором нарушается выведение ионов водорода или реабсорбция фильтрованного бикарбоната, что приводит к хроническому метаболическому нарушению гомеостаза с гипохлоремией. Наиболее распространенным вариантом является генерализованный тип заболевания, второе место занимает поражение дистальных канальцев нефронов, форма с патологией проксимальных канальцев встречается реже. Точный уровень заболеваемости отследить сложно, поскольку латентные формы часто не распознаются. Наследственные почечные тубулопатии встречаются реже приобретенных.
Причины
Основная причина патологии – утрата способности почек подкислять мочу. Существует множество состояний, которые вызывают ацидоз почечных канальцев, при одних заболеваниях поражаются дистальные отделы, при других – проксимальные, иногда патология носит сочетанный характер или вызывается рядом нарушений, связанных с альдостероном. Характерными причинами для каждого типа являются:
- Первый (классический). При врожденных состояниях кислотно-щелочное равновесие изменяют мутации генов ATP6V1B1, ATP6V0A4 S1C4A, что инициирует дистальный почечный тубулярный ацидоз (синдром или болезнь Батлера-Олбрайта). Вторичная форма становится результатом приема лекарств, трансплантации почек и некоторых заболеваний (серповидноклеточной анемии, системной красной волчанки, синдрома Шегрена). Медуллярная губчатая почка, цирроз и хроническая обструктивная уропатия также рассматриваются как потенциальные триггеры.
- Второй (проксимальный). Проксимальный почечный канальцевый ацидоз сопровождает синдром Фанкони, нефропатию на фоне множественной миеломы, как осложнение развивается при длительном приеме ацетазоламида, сульфонамидов, ифосфамида, тетрациклина. Реже имеет другую этиологию: дефицит витамина Д, хроническую гипокальциемию с вторичным гиперпаратиреозом, ряд наследственных патологий: непереносимость фруктозы, окулоцереброренальный синдром, цистиноз.
- Четвертый (обобщенный, генерализованный). Почечный тубулярный ацидоз обусловлен дефицитом альдостерона или нарушением его взаимодействия с рецепторами. Как первопричину нарушения рассматривают прием калийсохраняющих диуретиков, циклоспорина, гепарина, других препаратов. Из нефрологических заболеваний тубулопатию провоцируют ХБП, хронический интерстициальный нефрит, ВИЧ-инфекция с поражением почек, в урологии – все ситуации, связанные с обструкцией мочевыводящих путей. Тубулярный почечный ацидоз (IV) вызывает аутоиммунная дисфункция (СКВ), серповидноклеточная анемия, диабет.
К утяжеляющим факторам относят диарею, отравление солями тяжелых металлов, оперативные вмешательства на органах ЖКТ с установлением дренажей, которые выводят основания вместе с жидким содержимым или секретом (например, поджелудочной железы). Длительная диарея и увеличение объема стула приводят к дополнительной потере бикарбонатов. Прием некоторых лекарственных препаратов – кальция хлорида, магния сульфата, холестирамина нарушают гомеостаз, в сочетании с почечной патологией усугубляют состояние.
Патогенез
При первом типе имеет место сбой секреции или всасывания ионов водорода в дистальных канальцах, что вызывает высокую кислотность крови с гиперхлоремией. При этом нарушается способность почек поддерживать нормальный градиент концентрации водородных ионов между кровью и тубулярной жидкостью. Гипотетически это может быть связано с уменьшением активности клеток, секретирующих ионы водорода, дефектом энергетического механизма, дефицитом систем транспортировки, патологическими изменениями А и Р-клеток нефрона, ответственных за выработку Н+. Гиперкальциурия, сниженная экскреция цитратов приводит к нефролитиазу.
При II типе присутствует дисфункция проксимальных трубочек с нарушением процессов реабсорбции бикарбонатов, НСО3-, но ацидогенетическая функция дистальных структур нефрона остается сохранной. Патогенез проксимального тубулярного ацидоза объясняют дефицитом или полным отсутствием карбоангидразы И-(С), снижением активности карбоангидразы 1-(В) крови и/или митохондриальной НСО3-АТФ-азы в мембранах эпителия проксимальных канальцев. Механизмы развития остеомаляции и остеопении (в том числе, рахита у детей) включают гиперкальциурию, гиперфосфатурию, дисметаболизм витамина Д.
Тип IV инициирует дефицит альдостерона или невосприимчивость к нему дистальных канальцев. Характерен дисбаланс электролитов: натрия, калия, хлора и гидрокарбоната. Гипокалиемия способствует уменьшению секреции аммиака, приводя к метаболической ацидемии. Это расстройство – наиболее распространенный вариант почечной дистальной тубулопатии – имеет вторичный или спорадический характер по отношению к нарушению оси ренин-альдостерон-ренальной системы.
Классификация
- Первый (тубулярный классический, дистальный). Заболевание вызвано уменьшением выработки молекул водорода дистальными канальцами, повышением экскреции HCO3- . Часто сопровождается нефрокальцинозом, камнеобразованием. Выделяют врожденный (аутосомно-доминантный, аутосомно-рецессивный с тугоухостью, аутосомно-рецессивный без нарушения слуха) и приобретенный варианты.
- Второй (тубулярный проксимальный). Возникает из-за патологии проксимального отдела почечных трубок. Ацидемия менее выражена из-за сохранности дистальных интеркалированных клеток, вырабатывающих кислоту. Основной особенностью синдрома является деминерализация костной ткани (остеомаляция, рахит) из-за истощения фосфатного обмена. Различают аутосомно-рецессивный, аутосомно-доминантный, спорадический (транзиторный детский и персистирующий взрослый) почечный ацидоз.
- Третий (тубулярный ювенильный). Представляет собой сочетание классического и второго типа почечной ацидемии. Аутосомно-рецессивная патология, связанная с генетической мутацией. Регистрируется крайне редко. Тубулопатию поддерживает унаследованный дефицит углекислой ангидразы 2.
- Четвертый (тубулярный гиперкалиемический). Вызван дефицитом или резистентностью к альдостерону. Сопутствующая гиперкалиемия приводит к патологическим процессам в миокарде. Почечная функция не нарушена или страдает незначительно. Состояние встречается преимущественно у взрослых, в большинстве наблюдений имеет вторичный характер.
Симптомы
Симптомы вариативны, связаны с причинными факторами. При первичном генетически обусловленном дистальном почечном тубулярном ацидозе до появления развернутых клинических проявлений у детей обнаруживается бледность кожных покровов, мышечная гипотония, полиурия и, как следствие, полидипсия. В ряде случаев наследственную патологию сопровождает тугоухость, глазные аномалии. Температура тела незначительно повышена, есть склонность к запорам. Иногда единственным симптомом становится отставание в физическом развитии от сверстников.
В тяжелых случаях без лечения примерно по достижении 2-летнего возраста происходит деформация костей скелета. К 3-4 годам, если патология не диагностирована, и ребенок не получает лечения, прогрессирует задержка роста и интеллектуальных способностей. Кости нижних конечностей Х-образно искривляются, грудная клетка приобретает бочкообразную форму, голова выглядит непропорционально большой. Патологические переломы, костные боли, снижение мышечного тонуса свидетельствуют о прогрессировании патологического процесса.
Реакциями со стороны нервной системы при длительно существующих обменных нарушениях являются раздражительность, агрессивность. Типично раннее наступление пубертата. На фоне развившегося нефрокальциноза присоединяются рецидивирующие инфекции мочевыводящих путей, которые проявляются резями при учащенном мочеиспускании, поясничными болями, повышением температуры. Нефролитиаз и самостоятельное отхождение конкрементов сопровождаются почечной коликой, тошнотой, рвотой.
Проксимальный тип тубулярного почечного ацидоза имеет схожую симптоматику, но рахитоподобные изменения развиваются раньше, а нефрокальциноз выявляется значительно реже. Для второго варианта тубулопатии характерно более тяжелое течение, без лечения патология может прогрессировать до ацидемической комы. Типичен эксикоз (обезвоживание) с обмороками, резким цианозом, одышкой. Изменению подвергается волосяной покров: волосы колючие, жесткие, при соответствующей терапии становятся мягче. Иногда патологическое состояние проходит самостоятельно без какого-либо лечения к 3-10 годам.
У взрослых электролитные расстройства протекают бессимптомно или порождают более мягкую симптоматику. Гипокалиемия вызывает мышечную слабость, снижение рефлексов, параличи. Проявления со стороны опорно-двигательного аппарата включают остеомаляцию, боли. Подобные признаки более типичны для проксимального почечного ацидоза, но могут определяться и при дисфункции дистальных канальцев. Четвертый тип чаще бессимптомный, диагностируется при выявлении незначительной ацидемии. Существует риск нарушений сердечного ритма, особенно если состояние сопровождается выраженной гиперкалиемией.
Осложнения
Без надлежащего лечения хроническая кислотность крови осложняется задержкой роста, нефролитиазом, переломами. У пациентов наблюдают рецидивирующий пиелонефрит, нефрокальциноз, постепенную утрату почечной функции вплоть до терминальной стадии ХПН. Основным последствием дистальной тубулопатии является гипокалиемия, которая сопровождается аритмией, нередко становящейся причиной летального исхода. У детей отсутствие терапии ведет к тяжелым деформациям костей, задержке психического развития. Выраженная гиперкалиемия (IV) осложняется перепадами АД, бронхоспазмом, кишечными коликами.
Диагностика
Ведущая роль в диагностике принадлежит лабораторным анализам. Инструментальные исследования назначаются для определения основной патологии при вторичном почечном тубулярном ацидозе. При наличии в роду наследственных заболеваний необходима консультация генетика, при признаках обструктивной нефропатии требуется осмотр уролога. У детей с наследственными формами, сопровождающимися врожденной глухотой, в обследовании принимает участие сурдолог. Алгоритм диагностики индивидуален, результаты анализов и инструментальных методов вариативны.
Инструментальная диагностика включает УЗИ почек и мочевого пузыря, КТ и МРТ для исключения обструктивных уропатий, нефролитиаза. При тубулярном ацидозе (I) на сонограммах всегда определяется патология почек, для проксимального типа она нехарактерна. На рентгенограммах у детей визуализируются рахитические изменения скелета, поражение трубчатых костей голеней, кистей. Минеральную плотность костной ткани определяют с помощью денситометрии. Деминерализация костей без явного рахита или остеомаляции сопутствует 1 и 4 типам, витамин Д-ассоциированный рахит, остеомаляция характерны для 2 варианта.
Дифференциальную диагностику проводят между возможными типами нарушения кислотно-щелочного равновесия, почечными патологиями иного генеза. Синдром Фанкони имеет схожие проявления с проксимальным и дистальным тубулярным ацидозом. Генетически обусловленные формы дистального почечного ацидоза различают с псевдогипоальдостеронизмом, первичной гипероксалатурией, нефрокальцинозом иного генеза. Иногда требуется дифференциация с почечной недостаточностью, сахарным диабетом, салицилатной нефропатией.
Лечение почечного тубулярного ацидоза
Лечение направлено на коррекцию дисбаланса электролитов (калия, кальция, фосфатов), предотвращение осложнений, коррелирует с типом патологии. Операции выполняют только для исправления тяжелых деформаций костей. Основным мероприятием остается назначение ощелачивающей терапии, без лечения при тубулярном почечном ацидозе у детей замедляется рост, страдает умственное развитие. Щелочные агенты помогают достичь относительно нормальной концентрации бикарбонатов плазмы, скорректировать нарушения.
Медикаментозная терапия
Лечение классического почечного тубулярного ацидоза необходимо для поддержки роста у детей, ликвидации костных изменений, профилактики накопления интерстициальной тканью кальция. Назначают бикарбонат натрия, калия-натрия гидроген цитрат. При остеопатии используют активные метаболиты витамина Д3, что требует осторожности из-за риска развития гиперкальциурии. Терапия проксимального варианта включает пероральное введение цитрата калия. При резистентности в схему добавляют гидрохлортиазид. При ювенильном варианте применяют цитрат натрия или цитрат калия. Дозы подбирают индивидуально.
Основное лечение четвертого варианта направлено на восполнение или замещение альдостерона. Пациентам рекомендуют исключить из питания продукты, богатые калием. Дополнительно вводят NaCl на фоне мониторинга К крови, альдостерона, ренина плазмы. Необходимо прекратить прием лекарств, поддерживающих гиперкалиемию, осуществлять лечение основной патологии (ЗГТ минералокортикоидами). Назначают диуретики (фуросемид, гидрохлортиазид). При отсутствии ответа на лечение используют флудрокортизон, бикарбонат натрия для компенсации ацидемии и усиления экскреции К.
Прогноз и профилактика
Своевременно начатое лечение улучшает исход для всех случаев. При дистальной тубулопатии прием препаратов до присоединения нефрокальциноза благоприятно влияет на прогноз. Проксимальная транзиторная форма с возрастом может купироваться самостоятельно, но подщелачивающая терапия обеспечивает нормальный рост и препятствует деминерализации, рахиту, у взрослых лечение предотвращает развитие остеомаляции. Прогноз почечного ацидоза (IV) определяется тяжестью основного заболевания. За пациентами наблюдают, контролируют электролиты крови каждые 12 недель, проводят УЗИ почек 1 раз в 6 месяцев.
Меры профилактики для врожденной патологии отсутствуют, при отягощенном семейном анамнезе показана консультация генетика. Важно своевременное лечение урологических и нефрологических заболеваний, которые могут приводить к тяжелым формам тубулопатий. Особого наблюдения требуют пациенты с почечным канальцевым ацидозом на фоне уремии (почечная недостаточность с нарушением скорости клубочковой фильтрации, высоким уровнем креатинина, мочевины). Больные, получающие адекватное лечение, обычно не имеют выраженных симптомов, многие из них могут вести нормальную жизнь.
1. Федеральные клинические рекомендации по оказанию медицинской помощи детям с тубулопатиями/ Баранов А.А. – 2015.
2. Ренальный тубулярный ацидоз (обзор литературы)/ Вашурина Т.В., Сергеева Т.В.// Нефрология и диализ – 2003 - №2.
Почечный канальцевый ацидоз
Почечный канальцевый ацидоз. Проксимальноканальцевый
Почечный канальцевый ацидоз — это метаболический ацидоз с нормальным анионным интервалом. В его основе лежит нарушение либо реабсорбции бикарбоната, либо экскреции кислот (водородных ионов). Существуют как врожденные, так и приобретенные первичные и вторичные формы этой патологии. Различают три основные формы почечного канальцевого ацидоза: проксимальноканальцевый (II тип), дистальноканальцевый (I тип) и гиперкалиемический (IV тип).
Некоторые авторы выделяют и III (смешанный) тип почечного канальцевого ацидоза, при котором имеются элементы ацидоза как I, так и II типа. Он наблюдается главным образом у больных с наследственной недостаточностью карбоангидразы.
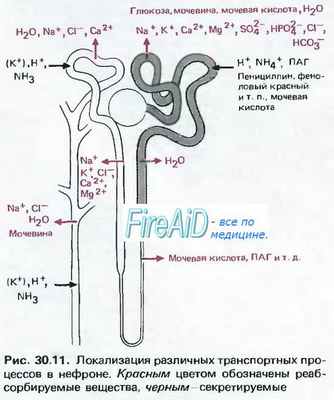
Нормальное закисление мочи. Закисление мочи определяется двумя процессами: реабсорбцией бикарбоната и экскрецией ионов водорода. Реабсорбция бикарбоната, приводя к уменьшению его содержания в клубочковом фильтрате, сама по себе не означает усиления секреции кислоты. Примерно 85 % фильтруемого бикарбоната реабсорбируется в проксимальных канальцах. У грудных детей этот процесс менее эффективен, поэтому почки могут выводить бикарбонат даже при его концентрации в сыворотке менее 22 ммоль/л.
В канальцах не существует специального транспортера бикарбоната, его реабсорбция осуществляется непрямым способом. Проксимальные канальцы секретируют ионы водорода в обмен на ионы натрия. Водородный ион связывается в просвете канальца с бикарбонатом, который под действием карбоангидразы превращается в двуокись углерода и воду.
Затем СО2 диффундирует в клетки проксимальных канальцев, где в результате ряда химических реакций вновь образуются молекулы бикарбоната (поступающие в околоканальцевые капилляры) и водородные ионы, которые участвуют в дальнейшей нейтрализации бикарбоната в просвете канальца. Оставшиеся 15 % бикарбоната реабсорбируются в дистальных отделах нефрона. В процессе обмена веществ в организме образуется около 1 ммоль/кг/сут кислоты, которая выводится путем секреции водородных ионов (за счет действия Н+-АТФазы вставочных клеток собирательных трубочек), а также ионов аммония и титруемых кислот (образующихся при взаимодействии ионов Н+ с органическими анионами, например фосфатом).
Проксимальноканальцевый ацидоз развивается при нарушении реабсорбции бикарбоната в проксимальных канальцах. Встречаются, хотя и редко, его изолированные врожденные или приобретенные формы. Описана изолированная аутосомно-доминантная форма и аутосомно-рецессивная с нарушением зрения. Гораздо чаще проксимальный канальцевый ацидоз отражает общую дисфункцию проксимальных канальцев, как при синдроме Фанкони, который помимо ацидоза характеризуется низкомолекулярной протеинурией, глюкозурией, фосфатурией и аминоацидурией.
Встречаются как аутосомно-доминантные, так и аутосомно-рецессивные формы первичного синдрома Фанкони. Кроме того, синдром Фанкони может быть и вторичным при некоторых врожденных нарушениях почечных канальцев или приобретенных заболеваниях. Многие из них являются наследственными. У детей синдром Фанкони часто развивается при лечении нефробластомы и других солидных опухолей фосфамидом.
Учебное видео анализа КЩС при респираторном и метаболическом ацидозе
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: