Прионы. Прионовые инфекции. Патогенез прионовых инфекций.
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Прионовые болезни – это прогрессирующие нейродегенеративные заболевания с летальным исходом. Механизм возникновения прионовых болезней изучен не до конца, однако в настоящее время считается, что заболевание развивается при накоплении в клетках центральной нервной системы избыточного количества патологического прионного белка.
Прионовые болезни встречаются очень редко – ежегодно регистрируется один случай на миллион человек. На данный момент известны 5 разновидностей этой патологии – спорадическая болезнь Крейцфельда-Якоба, новый вариант болезни Крейцфельда-Якоба, куру, синдром Герстманна-Штройслера-Шейнкера и фатальная семейная инсомния. Наиболее широко распространена спорадическая болезнь Крейцфельда-Якоба.
Болезнь может возникать попадании в организм патологического прионного белка извне (от человека или животного), иметь наследственную природу или возникать спонтанно как следствие генетических аномалий de novo. Течение определенных вариантов зависит от типа прионного белка. Инкубационный период может достигать 15 лет. Продолжительность жизни пациентов с прионными болезнями чаще всего составляет около года, реже – чуть более двух лет. Лечениесимптоматическое.
Синонимы русские
Трансмиссивная губчатая энцефалопатия.
Синонимы английские
Human prion diseases, transmissible spongiform encephalopathies, TSE.
Симптомы
Симптомыпатологии могут возникнуть в период от полугода до 10-15 лет после заражения. Проявления болезни могут развиваться постепенно, с неспецифических симптомов – бессонницы, вялости, апатии, заторможенности. В ряде случаев болезнь начинается внезапно и может напоминать делирий. Наиболее частыми проявлениями прионовых болезней являются:
- Прогрессирующая деменция
- Нарушение зрения
- Нарушение координации
- Нарушение речи
- Тремор
- Ригидность мышц
- Миоклонии
- Депрессия
- Тревожность
- Эмоциональная лабильность
Все проявления прионных болезней неуклонно прогрессирую, приводя в конечных стадиях болезни к акинетическому мутизму и коме.
Общая информация о заболевании
Прионные болезни встречаются как у человека, так и среди животных. Патогенез прионных болезней связывают с накоплением в организме патологического прионного белка. Прионный белок в норме присутствует в клетках организма животных и человека – это так называемый нормальный прионный белок. Он находится в наибольших количествах в нейронах и частично в клетках лимфоидной ткани. Прионный белок устойчив к высоким температурам, радиации, действию протеаз и химических веществ – алкоголя, формалина и других. Функции прионных белков неизвестны. Информация о структуре этих полипептидов закодирована в коротком плече 20 хромосомы у человека.
При изменении конфигурации нормальный прионный белок может превращаться в патологический. Патологический прионный белок, в свою очередь, способен запускать механизм преобразования нормального прионного белка в патологическую форму. Патологический прионный белок может возникать в организме спонтанно. В некоторых случаях накопление патологического прионного белка связано с наследственными генетическими нарушениями. На данный момент известно порядка 30 вариантов генетических нарушений, в результате которых меняется структура прионного белка. В ряде случае прионные болезни носят инфекционную природу. Это означает, что патологический прионный белок может попасть в организм человека извне – при пересадке роговицы, твердой мозговой оболочки, нейрохирургических операциях. В данном случае большое значение имеет устойчивость прионов к любым видам стерилизации. Известны случаи заражения патологоанатомов. Считается, что существует вероятность заражения человека при употреблении мяса коров, зараженных коровьим бешенством.
Несмотря на то, что часть прионных заболеваний имеет инфекционную природу, прионы отличаются от других инфекционных агентов – в их составе отсутствуют нуклеиновые кислоты (ДНК или РНК). Это осложняет процесс изучения патогенеза прионовых болезней. Известно, что накопление патологического прионового белка приводит к разрушению клеток нервной системы, мультифокальным спонгиоформным (губкоподобным) повреждениям тканей нервной системы, астроглиозу при отсутствии признаков воспалительной реакции. В связи с устойчивостью к действию протеаз, патологический прионный белок не может быть выведен из организма. Около 85 % пациентов погибают в течение года после появления первых симптомов болезни.
Кто в группе риска?
- Люди определенных профессий (медицинские работники, ветеринары, работники скотобоен, зоологи)
- Люди, перенесшие хирургические вмешательства, в том числе гемотрансфузии
- Люди, среди родственников которых были выявлены случаи наследственных прионных болезней
- Люди, употребляющие плохо термически обработанное мясо
Диагностика
К сожалению, на данный момент диагностика прионных болезней возможна лишь после возникновения клинических проявлений, то есть на этапе, когда болезнь зашла уже достаточно далеко. Раннее, пресимптоматическое выявление данной патологии невозможно. Схема диагностики прионных болезней не разработана окончательно.
Прионы. Прионовые инфекции. Патогенез прионовых инфекций.
Научный центр неврологии РАМН, Москва
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2012;112(9‑2): 59‑63
Стойда Н.И., Завалишин И.А. Прионные болезни. Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2012;112(9‑2):59‑63.
Stoĭda NI, Zavalishin IA. Prion diseases. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2012;112(9‑2):59‑63. (In Russ.).
Прионные болезни (ПБ) - группа нейродегенеративных заболеваний, вызываемых инфекционными белками - прионами. Известно 4 заболевания, обусловленные этим возбудителем: болезнь Крейтцфельдта-Якоба, фатальная инсомния, синдром Гертсманна-Штреусслера-Шейнкера и куру, которые могут проявляться в виде спорадических, инфекционных и наследственных форм. Приводятся данные клинических проявлений, патогенеза и патологической анатомии этих заболеваний. Обсуждаются вопросы диагностики, лечения и профилактики.
Научный центр неврологии РАМН, Москва
Прионные болезни (ПБ) - группа нейродегенеративных заболеваний человека и животных, вызываемых инфекционными белками - прионами.
Известно 4 болезни человека, вызываемые прионами - болезнь Крейтцфельдта-Якоба (БКЯ), куру, синдром Гертсманна-Штреусслера-Шейнкера (СГШШ) и фатальная инсомния (ФИ) [3].
Перечисленные болезни могут манифестировать в виде спорадических, инфекционных и наследственных форм. К группе приобретенных ПБ относится куру, регистрируемая в одном из племен Папуа - Новой Гвинеи, возникшая в результате употребления в пищу мозга умерших соплеменников во время ритуального каннибализма; ятрогенная БКЯ, развивающаяся в связи со случайным заражением пациента прионами; а также вариант БКЯ, возникновение которой связано с эпизоотией так называемого коровьего бешенства в Англии в 90-х годах XX века, возбудителем которого является прион. Группа спорадических ПБ включает идиопатические БКЯ и ФИ. Семейная БКЯ, СГШШ и семейная ФИ являются доминантно наследуемыми ПБ, которые связаны с мутацией прионного гена (PRNP), локализованного на коротком плече 20-й хромосомы.
Общая годовая частота спорадической формы БКЯ в разных регионах мира практически одинакова и не превышает 1,5-2 случая на 1 000 000 населения [7]. СГШШ регистрируется с частотой 1 случай на 1 000 000 населения, описано всего около 100 случаев ФИ, куру имеет место лишь в одной небольшой популяции.
Причиной ПБ является патологический прионный белок, причем его появление связано с соматической мутацией прионного гена (PRNP) или спонтанной конверсией нормального прионного белка (PrP с ) в его патологическую форму (PrP Sс ) при спорадических ПБ, мутацией PRNP и инвазией PrP Sс соответственно при наследственных и приобретенных формах этой патологии. Накопление PrP Sс в последующем связывается с его способностью трансформировать PrP с в его инфекционную форму за счет его (нормального белка) конформационных (т.е. пространственных) изменений. PrP Sс отличается от PrP с высокой резистентностью к нагреванию, ультрафиолетовому и рентгеновскому облучению, а также к протеазе К. Последнее относится к фрагменту PrP Sс 27-30 (PrP res ), причем выделяется три его типа (1, 2А, 2В), ассоциирующихся с определенным фенотипом ПБ. Прионный белок контагиозен независимо от причин его возникновения.
В результате в большинстве экспериментальных исследований установлено, что патогенез ПБ формируется в два этапа: экстрацеребральный и церебральный. На первом этапе вслед за интрацеребральной, интраперитонеальной или оральной инвазией PrP Sс поступает в органы лимфоретикулярной системы, где реплицирует. Последующий перенос PrP Sс в центральную нервную систему (ЦНС) опосредует автономная нервная система. Однако часть инфицированных лимфоцитов и макрофагов может проникать в ЦНС через гематоэнцефалический барьер (ГЭБ) непосредственно. PrP Sс может концентрироваться в хронических очагах воспаления, сопровождающихся развитием лимфоидных фолликул в различных органах, в том числе и секретирующих (печень, почки, поджелудочная железа, молочная железа и др.); причем в секретах последних также были обнаружены прионы [8]. На церебральном этапе главным является нарушение деградации PrP Sс в нейроне в связи с его конформационным отличием от PrP с и приобретение им нейротоксических свойств.
Важным фактором, определяющим вариабельность клинических и гистологических фенотипов всех ПБ, является генетический полиморфизм 129 кодона PRNP, кодирующего метионин и/или валин, а также штамм возбудителя [5, 9].
При гистологическом исследовании головного мозга при всех ПБ определяется гибель нейронов, спонгиоз, пролиферация астроцитов, амилоидные бляшки.
Спорадическая БКЯ отмечается в 90% от всех случаев болезни, остальные 10% регистрируются как наследственные и приобретенные. Соотношение мужчин и женщин составляет 1,5:1. Наследственная предрасположенность к этой форме ПБ связывается с генетическим полиморфизмом в 129-м кодоне PRNP. Основными клиническими проявлениями БКЯ являются быстро прогрессирующая мультифокальная деменция, как правило, с миоклонусом, а также экстрапирамидные, мозжечковые и пирамидные нарушения. Заболевание обычно регистрируется в старшей возрастной группе, его пик приходится на 60-65 лет [6]. Среднее время выживания около 8 мес, 90% больных умирают в течение первого года болезни.
В соответствии с данными литературы и собственными исследованиями можно выделить 5 стадий течения спорадической БКЯ:
1. Продромальная стадия (астения, адинамия, общая слабость, головокружения, головная боль, нарушения сна, боли в ногах, снижение аппетита, потеря массы тела, изменения в поведении, нарушения внимания и памяти).
2. Стадия первых симптомов (быстро нарастающие психические нарушения, зрительные и глазодвигательные нарушения, атаксия, дизартрия, скованность в ногах, дрожание в руках, галлюцинации, нарушения мочеиспускания).
3. Развернутая стадия (деменция, пирамидно-экстрапирамидные и мозжечковые нарушения, миоклонии, головокружения, зрительные и глазодвигательные нарушения, вегетативные расстройства, атрофии мышц).
4. Финальная стадия (деменция, акинетический мутизм, расстройства сознания, децеребрационная ригидность, миоклонии, сопутствующая соматическая патология, трофические нарушения, нарушения дыхания центрального типа, которые и являются причиной гибели этих пациентов).
5. Стадия продленной жизни (отсутствие собственного дыхания - больной находится на ИВЛ, апаллический синдром, вегетативный статус, гиперкинезы, контрактуры суставов, потеря мышечной массы, полипатия, причина смерти - сердечная недостаточность в течение ближайших нескольких месяцев).
Из всех рутинных методов диагностики значение имеет только электроэнцефалография.
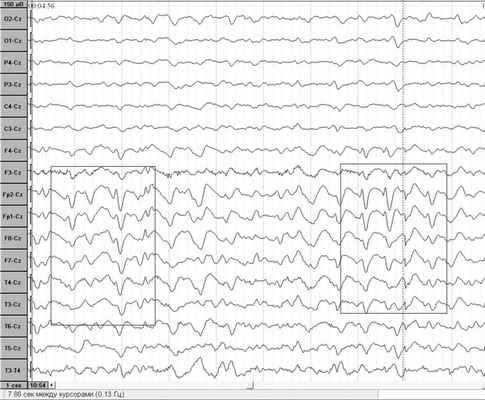
Изменения на ЭЭГ наблюдаются в развернутой стадии заболевания в виде двух- или трехфазных острых волн с частотой 1-2 в секунду, которые обычно накладываются на общий сниженный фон активности (см. рисунок). Рисунок 1. ЭЭГ больного БКЯ, развернутая стадия. В последние годы у этих больных в цереброспинальной жидкости (ЦСЖ) обнаружен атипичный белок 14.3.3, которому придается диагностическое значение [2, 4]. При МРТ-исследовании головного мозга этих больных может определяться усиление ответа от подкорковых ядер [1].
При дифференциальной диагностике БКЯ следует учитывать неврологические осложнения системных васкулитов, нейросифилис и криптококковый менингоэнцефалит; группу заболеваний, проявляющихся миоклонус-эпилепсией, мнестико-интеллектуальными нарушениями и атаксией (митохондриальная энцефаломиопатия с синдромом «рваных» красных волокон, сиалидозы, болезнь Лафора, болезнь Унверрихта-Лундборга, нейрональный цероидный липофусциноз). Начальные проявления комплекса СПИД-деменция могут напоминать БКЯ по началу заболевания, так же как и болезнь Паркинсона, прогрессирующий надъядерный паралич, сосудистая энцефалопатия и вариант паранеопластического процесса.
По результатам морфологических исследований и по данным биопсии можно выделить 3 этапа формирования патологического процесса при БКЯ: на первом относительно раннем этапе болезни патологические изменения, особенно спонгиоз, выражены умеренно и имеют очаговый ограниченный характер (по данным прижизненной биопсии головного мозга). На втором этапе, при естественном течении болезни изменения в головном мозге выражены больше и они имеют диффузный характер, при этом отдельные нейроны выглядят неизмененными. На третьем этапе, относящемся к продленной жизни больного, наблюдается терапевтически обусловленный патоморфоз за счет качественного и количественного изменения патологической анатомии болезни. Спонгиоформные изменения в мозговой ткани распространяются на белое вещество, где выявляется распад аксонов; наблюдается массивная гибель нейронов, а в оставшихся имеет место липопигментная дистрофия.
Спорадическая ФИ дебютирует в возрасте от 25 лет до 71 года (в среднем в 49 лет). Продолжительность болезни чаще от 1 года до 2 лет, но описаны случаи, когда пациенты погибали раньше (через 6-7 мес) или позже (через 30-33 мес). В клинической картине выявляются инсомния, вегетативные и двигательные нарушения, изменения циркадианных ритмов секреции гормонов. Для PrP Sc при спорадической форме ФИ характерен 2-й тип. При полисомнографическом исследовании отмечается отсутствие физиологического паттерна сна, исчезает его циклическая структура. Нередко отмечается симпатическая гиперактивность в виде высокого уровня адреналина и норадреналина в плазме крови, отмечается повышение температуры тела и артериального давления, при этом уменьшаются их циркадианные колебания; последнее отмечено и в отношении содержания гормонов гипоталамо-гипофизарной системы [1].
Проблема куру в настоящее время в основном представляет исторический интерес, поскольку в связи с запретом каннибализма в 1956 г. случаи этого заболевания в племени фора (Папуа-Новая Гвинея) регистрировались все реже и только у лиц, родившихся до этого запрета. Вместе с тем это обстоятельство свидетельствует о том, что инкубационный период куру может быть более 50 лет.
Все случаи ятрогенных ПБ относятся к БКЯ. Случайная передача БКЯ встречается как следствие различных хирургических и медицинских манипуляций. К ятрогенным путям передачи относятся использование недостаточно простерилизованных нейрохирургических инструментов, пересадка твердой мозговой оболочки и роговицы, использование гормона роста или гонадотропина, полученных из гипофизов умерших людей [11].
Вместе с тем существует мнение, что может быть повторная вспышка варианта БКЯ. При этом проводится аналогия с куру, которая может развиться даже через 50 лет после участия в пиршествах каннибалов, но при этом поражаются лица, гомозиготные по валину в 129-м кодоне, которые составляют более 30% популяции.
В настоящее время известно более 30 мутаций PRNP, которые обусловливают развитие наследственных ПБ. Однако фенотипические особенности мутации зависят от полиморфизма кодона 129. Этот кодон влияет на экспрессию мутантного гена, приводящую к разным клиническим и морфологическим фенотипам болезни.
Наследственная БКЯ в основном идентична спорадической по клиническим и морфологическим параметрам. При ней описано 7 точковых мутаций и 6 инсерций.
СГШШ в популяции регистрируется с частотой 1 случай на 1 млн населения. Болезнь начинается на 3-м или 4-м десятилетии жизни и продолжается несколько лет (в среднем 5 лет). Начальными симптомами являются мозжечковые нарушения; позже присоединяется деменция, которая иногда может и не проявляться. В развернутой стадии болезни преобладают мозжечковые симптомы, но в некоторых семьях ведущими признаками могут быть экстрапирамидные нарушения; в других - паралич взора, глухота и слепота. Характерно отсутствие сухожильных рефлексов на ногах при наличии разгибательных патологических знаков. Миоклонии редки.
К настоящему времени при СГШШ известно 7 точковых мутаций и инсерций PRNP [1]. Дифференциальный диагноз СГШШ проводится с оливопонтоцеребеллярной атаксией, гепатоцеребральной дегенерацией, рассеянным склерозом, семейной формой болезни Альцгеймера, метахроматической лейкодистрофией, болезнью Рефсума.
Достоверный диагноз БКЯ устанавливается с помощью стандартных патоморфологических методов и/или в соответствующих лабораториях с помощью дополнительных методов (PrP-иммунохимические методы, западный блоттинг и/или выявление скрепи-ассоциированных фибрилл). Остальные случаи БКЯ трактуются как вероятные или возможные.
Вероятный диагноз спорадической БКЯ может иметь место в случае прогрессирующей деменции, а также типичных изменений на ЭЭГ или продолжительности болезни менее 2 лет и позитивном тесте на белок 14.3.3. в ЦСЖ при наличии двух из следующих клинических признаков: миоклонуса, зрительных или мозжечковых нарушений, пирамидных или экстрапирамидных нарушений, акинетического мутизма. При возможной БКЯ имеются те же критерии, что и при вероятной БКЯ, но при отсутствии изменений на ЭЭГ и отрицательном тесте на белок 14.3.3.
Приобретенная БКЯ регистрируется в случае прогрессирующего мозжечкового синдрома у больного, получавшего экстракты тканей, содержащие гормоны гипофиза, а также при наличии в анамнезе факторов риска возникновения заболевания (например, при трансплантации твердой мозговой оболочки или роговицы).
Диагностика варианта БКЯ может быть вероятной при регистрации 5 из 6 следующих клинических синдромов: психические нарушения и парестезии на ранних этапах болезни; атаксия; хорея, дистония или миоклонус; деменция; акинетический мутизм. Вероятность диагноза варианта БКЯ становится большей при наличии следующих признаков: отсутствие потенциальной возможности ятрогенного воздействия, отсутствие мутации PRNP, отсутствие типичной ЭЭГ, начало болезни до 50 лет, продолжительность болезни не более 6 мес, рутинные исследования, исключающие альтернативный диагноз; при МРТ-исследовании выявляется усиленный сигнал в режиме Т2 от зрительных бугров. При летальном исходе необходимо патологоанатомическое исследование.
Семейная форма БКЯ регистрируется при достоверном или возможном диагнозе БКЯ плюс достоверный или возможный диагноз БКЯ у ближайшего родственника, а также в случаях нейропсихических нарушений плюс специфические для заболевания мутации PRNP. Диагноз СГШШ и семейной ФИ достоверен после патоморфологического исследования мозга, а также в случае сходных неврологических и психических нарушений у ближайшего родственника при наличии специфических для этих заболеваний мутаций прионного гена. Вместе с тем наличия семейного анамнеза и мутации PRNP не должно быть в случае спорадической ФИ.
Куру может быть диагностирована только в племени фора в Папуа-Новой Гвинее, несмотря на большое клиническое и патологоанатомическое сходство с новым вариантом БКЯ.
Не существует эффективной этиологической и патогенетической терапии ПБ [10]. На ранних стадиях применяется симптоматическая терапия, корригирующая поведенческие нарушения, расстройства сна и миоклонии (амфетамины, барбитураты, антидепрессанты, бензодиазепины, нейролептики), на поздних - поддерживающая терапия. С целью профилактики варианта БКЯ ограничено использование лекарственных средств, приготовленных из тканей коров, прекращено производство гормонов гипофиза животного происхождения, предпочтение отдается генно-инженерным препаратам. В ряде стран введены ограничения на трансплантацию твердой мозговой оболочки. В случае наследственных форм ПБ, очевидно, необходимо использовать методы генетического консультирования. Профилактика спорадической БКЯ, составляющей основную массу таких заболеваний, не разработана.
Обзор прионных заболеваний (Overview of Prion Diseases)
Прионные болезни представляют собой прогрессирующие дегенеративные, неизлечимые и в конечном итоге летальные поражения головного мозга.
Известные типы включают
Прионные болезни возникают в результате структурного изменения нормального поверхностного мембранного белка, который называют клеточным прионным белком (РгР C ), чья точная функция неизвестна. Белки прионов с неправильной конформацией называются прионами или скрепи PrP (PrP Sc от названия прототипической прионной болезни овец).
Прионы (PRP Sc ) являются патогенными и часто заразными. Они вызывают прионную болезнь благодаря
Ауторепликации: PrP Sc индуцирует конформационные превращения PrP C , с созданием дубликатов PrP Sc , которые при цепной реакции вызывают дальнейшее превращение PrP C в PrP Sc . Этот процесс преобразования прионного белка PrP Sc распространяется на различные области головного мозга.
Вызывая гибель нейронов
Нормальный PrP C водорастворимый и чувствительный к протеазе, но большой процент PrP Sc не растворяется в воде и обладает заметной устойчивостью к разрушению протеазой (аналогичный бета-амилоиду при болезни Альцгеймера, который PrP Sc напоминает), приводя к медленной, но стабильной клеточной аккумуляции и гибели нейронов. Постепенное накопление прионов вызывает глиоз и характерные вакуолярные (спонгиформные) гистологические изменения, приводящие к развитию деменции и другим видам неврологических расстройств. Симптомы и признаки приобретенной прионной болезни формируются на протяжении многих месяцев и лет после первоначального заражения PrP Sc .
Прионные заболевания следует исключать у пациентов с деменцией, особенно если она быстро прогрессирует.
Механизм передачи прионных заболеваний
Возникает прионная болезнь
Спорадически (по-видимому, начинаясь спонтанно, без известной причины)
Вследствие генетического наследования (семейные)
Через инфекционную передачу
Спорадические прионные болезни являются наиболее распространенными, с ежегодной заболеваемостью в мировом масштабе 1 случай на 1 млн населения. Как с самого начала образуется PrP Sc неизвестно.
Семейные прионные заболевания вызваны дефектами в гене PrP, который содержится в коротком плече хромосомы 20. Генетические мутации, вызывающие прионные болезни являются аутосомно-доминантными, т.е. они могут вызывать болезнь при наследовании только от одного родителя. Кроме того, пенетрантность является вариабельной, т.е., в зависимости от типа мутации процент вариабельности носителей мутации имеют клинические признаки заболевания в течение их жизни.
Существует более 50 мутаций. Различные генные дефекты вызывают разные виды прионных заболеваний, которые включают
Семейная болезнь Крейтцфельдта-Якоба (БКЯ)
Синдром Герстманна–Штройсслера–Шейнкера (ГШШ);
Заболевания со смешанными признаками болезни Крейтцфельда-Якоба (англ. CJD) и болезни Герстманна-Штреусслера-Шейнкера (англ. GSS)
Заболевания, которые клинически и патогенетически отличаются от других прионных заболеваний, таких как прионная болезнь, связанная с диареей и вегетативной нейропатией
Фатальная семейная бессонница (FFI)
На сегодняшний день исследователи определили только одну мутацию, которая вызывает фатальную семейную бессонницу (ФСБ), наследственную форму фатальной бессонницы.
Мутации гена PrP изменяют аминокислотную последовательность PrP C , приводя к неправильной конформации белка и образованию белка PrP Sc . Небольшие аномалии в определенных кодонах (нуклеотидные последовательности, которые являются строительными блоками генов), которые сами по себе не вызывают заболевания, могут определять преобладающие симптомы и скорость прогрессирования заболевания при семейных и других прионных заболеваниях ( 1 Справочные материалы по механизмам передачи Прионные болезни представляют собой прогрессирующие дегенеративные, неизлечимые и в конечном итоге летальные поражения головного мозга. Известные типы включают Болезнь Крейтцфельда–Якоба (БКЯ). Прочитайте дополнительные сведения ).
Инфекционно передаваемые прионные заболевания являются редкими. Они могут передаваться
От человека к человеку: ятрогенно, при пересадке органов и тканей, при использовании загрязненных хирургических инструментов или, реже, при переливании крови (как в случае заражения вБКЯ); или с помощью каннибализма (как в случае заражения куру)
От животного к человеку: после употребления зараженного мяса (как в случае заражения вБКЯ)
Для прионных болезней нет подтвержденных данных, насколько заразен случайный контакт человека с человеком.
Прионные болезни присущи многим млекопитающим (например, норкам, лосям, оленям, домашним овцам и крупному рогатому скоту) и могут передаваться между видами через пищевую цепь. Тем не менее, передача от животных к человеку наблюдается только при вБКЯ, после того как люди употребляли говядину, полученную от крупного рогатого скота, больного губкообразной энцефалопатией крупного рогатого скота (ГЭКРС или коровьего бешенства).
В нескольких западных штатах США, Канаде и сейчас Южной Кореи и Норвегии ( 2 Справочные материалы по механизмам передачи Прионные болезни представляют собой прогрессирующие дегенеративные, неизлечимые и в конечном итоге летальные поражения головного мозга. Известные типы включают Болезнь Крейтцфельда–Якоба (БКЯ). Прочитайте дополнительные сведения ) существуют опасения, что хроническая изнуряющая болезнь (ХИБ), прионовая болезнь лосей и оленей, может передаваться мясникам, людям, которые охотятся или принимают в пищу мясо зараженных животных. Хотя передача ХИБ от животных к человеку является маловероятной, недавние данные указывают на то, что барьеры между видами могут быть ослаблены, если ХИБ передается от животного к животному несколько раз (как это может произойти в дикой природе [ 3 Справочные материалы по механизмам передачи Прионные болезни представляют собой прогрессирующие дегенеративные, неизлечимые и в конечном итоге летальные поражения головного мозга. Известные типы включают Болезнь Крейтцфельда–Якоба (БКЯ). Прочитайте дополнительные сведения ]).
Справочные материалы по механизмам передачи
1. Pocchiari M and Manson J, editors. Human Prion Diseases. В Handbook of Clinical Neurology , edited by M Pocchiari, and J Manson. New York, Elsevier, 2018, vol 153, pp. 2–498.
2. Benestad SL, Mitchell G, Simmons M, et al: First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res 47 (1):88, 2016. doi: 10.1186/s13567-016-0375-4.
3. Barria MA, Telling GC, Gambetti P, et al: Generation of a new form of human PrP Sc in vitro by interspecies transmission from cervid prions. J Biol Chem 286 (9):7490–7495, 2011. doi: 10.1074/jbc.M110.198465.
Патогенез прионных заболеваний состоит из двух стадий — инфекционной и токсической
Рис. 1. a. Линии мышей с высоким уровнем PrP C умирают от прионной болезни быстрее, чем те, у кого уровень PrP C низок. По горизонтальной оси — дни после заражения, по вертикальной — процент выживших животных. b. После заражения уровень патогенных прионов у всех трех линий мышей резко возрастает, а затем достигает плато. Отличия между линиями только в том, сколько времени они проживают после того, как плато достигнуто. По горизонтальной оси — дни после заражения, по вертикальной — логарифм количества инфекционных частиц на грамм мозговой ткани. Стрелочками показано среднее время окончания инкубационного периода (то есть начало болезни). Изображение из обсуждаемой статьи в Nature
Патологический прионный белок, попадая в нейрон, заставляет неправильно складываться нормальные прионы; переродившиеся белки слипаются в смертоносные агрегаты и убивают клетку. Выяснилось, что количество плохих прионов растет до одного и того же уровня независимо от исходного количества хороших, зато следующая фаза болезни — фаза плато — тем короче (и тем быстрее наступает смерть), чем больше хороших прионов было в клетке.
Прионные заболевания (см. прионы) — один из самых коварных и загадочных типов нейродегенеративных болезней. Сюда относятся болезнь Крейтцфельдта–Якоба, от которой страдают люди; коровье бешенство, которое поражает крупный рогатый скот (и которое напугало человечество 20 лет назад, когда в Великобритании началась эпидемия этой болезни); скрейпи, от которой умирают козы и овцы; страшное наследственное заболевание человека под названием фатальная семейная бессонница и некоторые другие болезни.
Патогенез заболевания таков. В нормальном состоянии в нейроне содержится некий белок под названием PrP C . Это обычный, добропорядочный белок; в его структуре много альфа-спиралей, и поэтому он хорошо растворяется в воде. Работа его состоит в том, чтобы поддерживать качество миелиновой оболочки (которая играет у нейрона примерно ту же роль, что и изоляция у провода).
Но кроме «хорошего», безвредного PrP C существует еще одна — патологическая — форма того же белка под названием PrP Sc . Она неправильно сложена — вместо альфа-спиралей у нее много бета-конформаций, которые очень любят слипаться друг с другом, образуя нерастворимые агрегаты. Агрегаты эти губительны для клетки — они «забивают» ее внутреннее пространство, «душат» органеллы и в конце концов доводят клетку до смерти.
Но самое ужасное то, что, оказавшись в клетке, патологический, вредоносный PrP Sc заставляет «хороший» PrP C сложиться «по-своему», неправильным образом. В результате в клетке нарастает количество неправильно сложенных белков, которые слипаются в смертоносные агрегаты и убивают ее.
Нейроны гибнут; неправильно сложенные PrP Sc из мертвых клеток заражают живые и их тоже доводят до смерти. Всё это приводит к тяжелым поражениям определенных областей мозга, и через некоторое время организм погибает.
Патогенез всех прионных заболеваний состоит из длинного инкубационного периода (у человека он может длиться до 50 лет!), во время которого не проявляется никаких признаков болезни; вслед за ним идет короткая (обычно в несколько месяцев длиной) клиническая фаза, когда уже невозможно ничего сделать. Надо отметить, что у разных людей инкубационный период может занимать совершенно разное время. Что конкретно происходит на этих двух стадиях заболевания в нейронах и с чем связано столь резкое проявление болезни, до настоящего времени было изучено слабо.
Некоторое время назад было замечено, что в инбредных (генетически идентичных) линиях лабораторных мышей инкубационный период прионных заболеваний продолжается примерно одно и то же время и хорошо воспроизводится от эксперимента к эксперименту. Причем этот период длится тем дольше (а болезнь наступает тем позже!), чем ниже в нейронах мышей уровень прионного белка PrP C . Но как следует изучить этот феномен было довольно трудно, так как не было разработано методики, позволяющей достаточно точно измерить уровень PrP Sc в клетках во время инкубационного периода.
Однако в 2003 году такая методика была предложена. Исследователи из Института неврологии Университетского колледжа в Лондоне использовали ее, чтобы понять, что происходит в нейроне, зараженном неправильно сложенными прионными белками.
В своих экспериментах они использовали несколько инбредных линий мышей:
1. Prnp null (Prnp o/o ), не экспрессирующие PrP C вовсе.
2. Hemizygous Prnp null (Prnp +/o ), экспрессирующие половину нормального уровня PrP C .
3. FVB/N (Prnp +/+ ), экспрессирующие нормальный уровень PrP C .
4. Tg20, экспрессирующие уровень PrP C в восемь раз больше нормального.
Экспериментальные группы мышей каждой линии были заражены прионным заболеванием, а контрольные получили «пустой укол»; после этого исследователи регулярно измеряли уровень инфекционных частиц в нейронах подопытных животных и следили за их состоянием.
Первый полученный результат был предсказуем. Одна линия — Prnp o/o — оказалась для болезни неуязвима; три другие заболели, причем Tg20 умирали быстрее Prnp +/+ , а Prnp +/+ — быстрее Prnp +/o .
Но когда исследователи попробовали измерить уровень прионов в мозге больных мышей, то результаты оказались гораздо более интересными.
Prnp o/o , как и следовало ожидать, справились с введенными инфекционными частицами и уничтожили их — уже через десять дней после заражения никаких прионов в их мозге не выявлялось.
Зато для остальных трех линий мышей наблюдалась парадоксальная картина. Вначале уровень патогенных прионов резко (и почти с одной скоростью для всех трех линий) увеличивался (фаза роста); затем он останавливался и практически переставал меняться (фаза плато). Получилось, что разница между тремя линиями мышей не в том, сколько «вредных» прионных частиц находится у них в мозге (у всех было примерно одинаковое количество), а в том, сколько времени продолжается эта фаза плато. Tg20 умирали практически сразу после ее достижения, а Prnp +/o , наоборот, жили еще долго после этого (и некоторое время не проявляли никаких признаков болезни), несмотря на то, что уровень инфекционных частиц в их мозге был так же высок, как и у Tg20.
Выяснился еще один интересный момент. Оказалось, что уровень PrP C у мышей обратно пропорционален продолжительности фазы плато (рис. 2). Для фазы роста такой зависимости не было выявлено, хотя небольшие различия по ее продолжительности между тремя линиями всё-таки наблюдались.
Рис. 2. Продолжительность жизни мыши после того, как уровень патогенных прионов в ее мозге достиг плато, обратно пропорциональна уровню в ее мозге PrP C . По горизонтальной оси — экспрессия PrP (уровень экспрессии дикого типа принят за единицу), по вертикальной — величина, обратная продолжительности фазы плато (в днях –1 ). За окончание фазы плато принято окончание инкубационного периода и, соответственно, начало болезни. Изображение из обсуждаемой статьи в Nature
Результат, мягко говоря, неожиданный. Гораздо логичней было бы, если бы фаза роста коррелировала с количеством PrP C в клетках (чем больше вокруг «хороших» прионов, тем быстрее они превращаются в «плохие»), а фаза плато, наоборот, нет (тогда бы можно было объяснить ее появление тем, что количество «плохих» прионов в клетке достигло максимально возможного, и теперь они постепенно будут разрушать все вокруг).
Но то, что после достижения прионами фазы плато животные живут разное время, ставит всё с ног на голову. Эти результаты объяснить трудно, если только.
Если только не предположить, что перед нами неполная картина. Что если на самом деле есть не два, а три вида прионов? Один из них — PrP C — «хороший», он не наносит клетке вреда. Второй — PrP Sc — «плохой»; это только инфекционный агент, но не токсический, то есть хотя он размножается в клетке, вызывая перерождение PrP C , но сам по себе не вызывает ужасных последствий. И, наконец, третий — PrP L — «злой», токсический агент, который доводит клетку до смерти; появление его, по мнению авторов, связано с наличием в клетке PrP Sc .
Патогенез болезни тогда выглядит так: в клетку попадает патогенный PrP Sc ; окружающие молекулы PrP C меняют конформацию и сами превращаются в PrP Sc (на графике это выглядит как фаза роста); затем достигается максимально возможный уровень PrP Sc (начинается фаза плато), теперь уже PrP Sc запускает образование PrP L из PrP C (фаза плато продолжается). Длительность фазы плато обратно пропорциональна количеству в клетке PrP C — если его много, то она будет короткой, если мало — наоборот, длинной. Иными словами, болезнь состоит из двух стадий — инфекционной (когда «работает» PrP Sc ) и токсической (когда за дело берется PrP L ).
В пользу этого предположения говорит многое — прежде всего то, что, действительно, помимо двух хорошо изученных конформаций прионного белка существует еще множество слабоисследованных. (Вполне возможно, что на самом деле в патогенезе участвуют даже не три, а больше форм белка.) Два главных автора обсуждаемой статьи еще в 2007 году высказывали сходную теорию, и эти результаты служат отличным подтверждением их идей.
Однако возможны и другие интерпретации полученных данных. Может быть, всё дело просто в особенностях измерения концентрации PrP Sc (методика появилась недавно, и многие ее подводные камни еще не известны). Кроме того, показано, что PrP С служит рецептором для PrP Sc и за счет запускаемых в результате их взаимодействия каскадов реакций в клетке возникают токсические эффекты. Если дело в этом, то понятно, почему продолжительность фазы плато зависит от уровня PrP С — чем его больше, тем быстрее «работает» PrP Sc и тем быстрее доводит клетку до смерти (и тем меньше длится фаза плато). В любом случае, полученные результаты очень интересны и явно требуют дальнейших исследований.
Источник: Malin K. Sandberg, Huda Al-Doujaily, Bernadette Sharps, Anthony R. Clarke, John Collinge. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases // Nature. 24 February 2011. V. 470. P. 540–542.
Не бактерия и не вирус. Опасные белки - прионы
Инфекционные болезни могут развиваться не только из-за бактерий, вирусов и других хорошо известных врачам микроорганизмов - это ошеломляющее заявление в медицинской среде было сделано после того, как в 1982 году профессор неврологии и биохимии Стэнли Прузинер (США) обнаружил белковые соединения, способные вызывать заболевания. Открытие белков-прионов было настоящим прорывом в медицине, доказательством чему стало получение учёным Нобелевской премии в 1997 году.
Прионы: биологическая сущность, свойства, среда обитания таинственных молекул
До недавнего времени исследователи считали, что в составе любой «живой» субстанции должны быть молекулы ДНК или РНК - нуклеиновых кислот, обусловливающих способность вирусов, бактерий, грибов и прочих организмов размножаться. Однако открытие прионов полностью трансформировало это представление. Устойчивость к высоким температурам, к различным видам излучений, действию нуклеаз (ферментов, способных расщеплять нуклеиновые кислоты), отсутствие роста на питательных средах – такими необычными свойствами обладал ранее не известный возбудитель.
Белковые соединения с определённой конфигурацией, способные трансформироваться в патогенные и вызывать нейродегенеративные процессы в организме, были названы прионами. Термин «прион» (prion) предложил Стэнли Прузинер. Термин происходит от фрагментов английских слов protein (белок) и infection (инфекция). Прионы способны размножаться. Этот процесс более продолжителен по времени, чем размножение патогенных микроорганизмов, поэтому от момента попадания прионов в организм до клинических проявлений болезни может пройти несколько месяцев или лет.
Молекула приона в «нормальной» форме имеется на поверхности нервных клеток у каждого человека. Обычные молекулы белка, вступая в контакт с патологическими, сами превращаются в них, изменяя при этом собственную пространственную структуру. Что является пусковым механизмом подобной трансформации, до конца не известно. Из этого следует, что прион, выступая в роли инфекционного агента, заражает нормальные молекулы, вызывая «молекулярную эпидемию».
Токсичные белковые бляшки на клетке приводят к её гибели, а на месте погибшей клетки образуется пустота, которая заполняется жидкостью. Количество пустот в головном мозге с течением времени будет увеличиваться, пока он не превратится в «губку».
Как можно заразиться прионами?
На сегодняшний день выделяют следующие основные пути заражения инфекционным белком-прионом:
1. Трансмиссивный. В этом случае молекулы белка передаются от одного вида млекопитающего к другому - например, от инфицированной коровы или овцы человеку. Заражение происходит при употреблении в пищу мяса или молока заражённого животного, либо использовании его тканей (роговицы, препаратов крови и т.п.), применении во время оперативных вмешательств биологического шовного материала.
2. Наследственный. Заболевание развивается на фоне генетической мутации, затрагивающей область 20-й хромосомы. Несмотря на слабую изученность функционирования этого участка генома, достоверно известно его участие в синтезе нормального прионного белка. В случае генных мутаций вместо здорового приона образуется патологический, а это приводит к развитию болезней.
3. Спорадический. При этом аномальный белок появляется в организме спонтанно, без видимых причин.
Вне зависимости от способа появления аномальный белок может стать причиной заражения других людей.
Прионные заболевания: особенности течения, лечения, прогноз
Отличительной особенностью болезней, вызываемых прионами, является длительный инкубационный период - от 2-3 месяцев до нескольких десятилетий. Подавляющее большинство прионных заболеваний человека являются спорадическими и имеют семейный характер наследования.
Куру, синдром Герстманна-Штреусслера-Шейнкера, болезнь Крейтцфельдта-Якоба, скрэпи – прионы вызывают заболевания, сопровождаемые поражением центральной нервной системы. Для них характерны такие признаки как деменция (слабоумие), зрительные и мозжечковые нарушения. При этом у больного могут отмечаться двигательные расстройства, бессонница, галлюцинации, нарушение речи.
К сожалению, эффективных методов лечения прионных болезней на сегодняшний день нет, хотя учёные пытаются предотвращать переход нормального белка в аномальный. Пациентам назначается симптоматическая терапия с использованием противосудорожных средств для облегчения страданий. Прогноз пока неутешителен, так как все вышеперечисленные заболевания завершаются летальным исходом.
Перспективы
Недостаточная изученность проблемы прионов и прионных болезней способствует углублению исследований в этой области - учёные занимаются активным поиском средств борьбы с патогенными белками. Актуальность этого вопроса растёт в связи с возможностью возникновения «прионной эпидемии», например, из-за приёма лекарственных средств животного происхождения.
Раскрытие загадочных явлений, которыми окутаны прионы, возможно, поможет в понимании ряда серьёзных биомедицинских проблем человечества.
Читайте также: