Семейная дегенерация роговицы, патология обмена кальция и глухота
Добавил пользователь Skiper Обновлено: 30.01.2026
ГБУ "Уфимский НИИ глазных болезней" АН Республики Башкортостан
ГБУ «Уфимский НИИ глазных болезней» Академии наук Республики Башкортостан, ул. Пушкина, 90, Уфа, Россия, 450008
ГБУ «Уфимский НИИ глазных болезней АН РБ», ул. Пушкина, 90, Уфа, 450008, Российская Федерация
Результаты лечения пеллюцидной маргинальной дегенерации роговицы методом роговичного кросслинкинга
Журнал: Вестник офтальмологии. 2017;133(3): 58‑66
Цель — изучить клиническую эффективность роговичного кросслинкинга у пациентов с прогрессирующим течением пеллюцидной маргинальной дегенерации роговицы (ПМДР). Материал и методы. Проведено лечение 9 пациентов (16 глаз) с ПМДР методом стандартного роговичного кросслинкинга. Роговицу насыщали раствором «Декстралинк», облучали УФ-А мощностью 3 мВ/см2 в течение 30 мин. Срок наблюдения 12 мес. Результаты. Через 1 мес после лечения отмечалось незначительное снижение некорригированной (НКОЗ) и корригированной (КОЗ) остроты зрения с 0,08±0,03 и 0,4±0,15 до 0,06±0,02 и 0,3±0,07 соответственно за счет псевдохейза роговицы. Кератометрические показатели — средняя преломляющая сила роговицы, роговичный астигматизм и толщина роговицы — существенно не менялись. Демаркационная линия определялась в 56% случаев. К 3-му месяцу НКОЗ и КОЗ улучшились до 0,1±0,07 и 0,52±0,1 соответственно. Преломляющая сила роговицы уменьшилась на 2,0 дптр, роговичный астигматизм — на 0,7 дптр, составив 46,8±2,7 и 5,1±1,3 соответственно (р≤0,04). Толщина роговицы в центре уменьшилась в среднем на 29 мкм. Демаркационная линия определялась в 25% случаев. Через 6 мес КОЗ составила 0,58±0,13, при этом отмечено повышение на 2 строки в 31% наблюдений, а на 1 строку — в 56%. Преломляющая сила роговицы снизилась до 45,7±1,6 дптр, роговичный астигматизм — до 4,8±1,5 дптр. Демаркационная линия не определялась. Через 1 год существенных изменений средних величин оптометрических показателей по сравнению с показателями 6-месячного периода не наблюдалось. Заключение. Пеллюцидная маргинальная дегенерация роговицы представляет собой разновидность первичных эктазий, процесс, как правило, двусторонний с характерной клинической картиной и поздним началом. Проведение фотохимического кросслинкинга роговицы при прогрессирующем течении заболевания способствует улучшению оптометрических показателей и стабилизации процесса.
Пеллюцидная маргинальная дегенерация роговицы (ПМДР) — невоспалительное заболевание, характеризующееся истончением и выпячиванием роговицы, которые наблюдаются в ее нижнем отделе от 4 до 8 часов. Обычно истончение шириной около 2 мм в виде серпа (ленты) расположено на расстоянии 2 мм от нижнего лимба, отделено от него нормальной роговицей, не сопровождается васкуляризацией, образованием кольца Флейшера или стрий Фогта [1—4]. Зона выстояния роговицы также расположена в нижних секторах. Еще в 60-е годы прошлого столетия J. Francois и соавторы на основании гистопатологических исследований высказали мнение о том, что ПМДР является формой проявления кератоконуса, характеризующейся специфичной локализацией [5, 6]. При кератоконусе наибольшее истончение роговицы регистрируется в зоне максимальной протрузии, а при ПМДР, наоборот, над истонченной областью формируется выстояние роговицы. Является ли ПМДР проявлением кератоконуса, все еще дискутируется в научной литературе.
Дебютирует заболевание в молодом возрасте (20—40 лет), прогрессирует медленно. Часто встречается двустороннее поражение с характерным истончением, выстоянием и иррегулярностью роговицы. Заболевание возникает спорадически, этиология не ясна. При эктатических заболеваниях роговицы происходит выраженное снижение остроты зрения, развитие аметропий высоких степеней, что служит причиной зрительной и социальной дезадаптации пациентов.
Топографические исследования роговицы при ПМДР констатируют высокий иррегулярный обратный астигматизм, укручение роговицы по типу «клешни краба», локальное истончение нижнего сектора циркулярно лимбу [6, 7].
Лечение ПМДР представляет большие трудности. Традиционно слабые и средние стадии заболевания корректируют ношением жестких контактных линз (ЖКЛ), при их непереносимости — имплантацией роговичных сегментов, а в тяжелых случаях проводят послойную или сквозную кератопластику и др. [8, 9, 11, 12].
Цель исследования — изучить клиническую эффективность КР у пациентов с прогрессирующим течением ПМДР.
Материал и методы
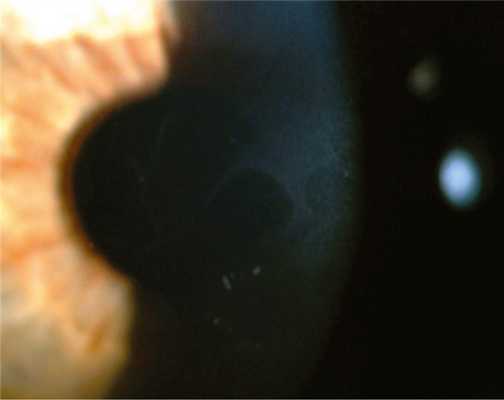
В исследование вошли 9 пациентов (16 глаз) с прогрессирующим течением заболевания, которым проведено лечение методом фотохимического кросслинкинга роговицы. Возраст пациентов составил от 29 до 58 лет. Выявлена характерная клиническая картина с истончением роговицы в нижней ее части шириной около 1—2 мм в 2 мм от лимба (рис. 1) и эктазией в виде «клешни краба» или «целующихся птичек». Толщина роговицы в месте истончения варьировала от 412 до 504 мкм (в среднем 468±36 мкм), величина средней преломляющей силы (Kave) — от 44,5 до 53,8 дптр (47,9±1,3 дптр), роговичного астигматизма — от 3,7 до 8,6 дптр (5,8±1,3 дптр), НКОЗ — от 0,04 до 0,3 (0,08±0,03), КОЗ — от 0,09 до 0,7 (0,4±0,15). Пациенты пользовались ЖКЛ, но с разной степенью толерантности: у 3 пациентов отмечалась непереносимость контактной коррекции, остальные испытывали дискомфорт и периодически прерывали ношение.

Рис. 1. Биомикроскопическая картина глаза пациента К. с пеллюцидной маргинальной дегенерацией роговицы. Истончение в нижнем секторе.
Кросслинкинг выполняли по стандартному методу, согласно Дрезденскому протоколу, с деэпителизацией роговицы в пределах 8 мм, насыщением роговицы раствором 0,1% рибофлавина и 20% декстрана («Декстралинк», Россия, рег. уд. №ФСР 2010/09071) в течение 15—20 мин и ультрафиолетовым облучением (370 нм) с помощью аппарата «УФалинк» (Россия, рег. уд. №ФСР 2009/05489) при мощности 3 мВ/см 2 в течение 30 мин. В послеоперационном периоде проводили местное противовоспалительное лечение антибиотиками, стероидными гормонами и слезозаменителями.
Процедуру выполняли с письменного согласия пациентов с информированием о преимуществах, недостатках, возможной эффективности лечения и включении результатов в научные исследования согласно Хельсинскому соглашению о правах человека. Всем пациентам проводили общепринятые и дополнительные офтальмологические методы исследования, включая кератотопографию (OPD—SCAN, «Nidek», Япония), оптико-когерентную томографию (ОКТ) переднего отрезка глаза и пахиметрию (Vizante ОСТ, «Carl Zeiss Meditec Inc.», Германия), конфокальную микроскопию при помощи роговичной насадки Rostok аппарата HRT—III («Heidelberg Retina Tomograph», Германия), исследование биомеханических свойств роговицы (Ocular Response Analyzer, «Reichert», США).
Критериями оценки эффективности лечения были уменьшение преломляющей силы роговицы на 1,0 дптр и более, улучшение остроты зрения на 1 строку и более по таблице Сивцева—Головина (Снеллена), наличие или отсутствие демаркационной линии (ДЛ).
Обследование проводили до и через 1, 3, 6 и 12 мес после лечения. Контактную коррекцию назначали не ранее, чем через 3 мес после процедуры.
Результаты
Во всех случаях проведения КР интра- и послеоперационных осложнений не наблюдалось. Роговичный синдром исчезал после полной эпителизации роговицы в течение 1—5 сут. В первые 2—4 нед наблюдалось развитие псевдохейза, обусловленное апоптозом кератоцитов.
Через 1 мес после лечения в половине случаев отмечалось снижение НКОЗ и КОЗ в среднем на 0,02±0,01 и 0,1±0,08 соответственно, что было связано с псевдохейзом. На рис. 2 представлена кератотопограмма пациента через 1 мес после процедуры, демонстрирующая уплощение и выравнивание передней поверхности роговицы и снижение ее преломляющей силы. Различия оптометрических показателей роговицы по сравнению с дооперационными были статистически малозначимы. Средний показатель преломляющей силы роговицы составлял 47,4±4,5 дптр, роговичного астигматизма — 5,7±2,5 дптр, толщина роговицы увеличилась до 480±130 мкм.

Рис. 2. Кератотопограмма пациента до (а), в динамике через 6 мес (б) и через 1 мес (в) после кросслинкинга. Снижение преломляющей силы роговицы с относительным выравниванием ее передней поверхности.
Через 3 мес НКОЗ и КОЗ повысились во всех случаях и составили 0,1±0,07 и 0,52±0,1 соответственно, в основном за счет снижения величины цилиндрического компонента рефракции. Отмечено уменьшение толщины роговицы в центре в среднем на 29 (439±153) мкм, преломляющей силы роговицы (Kave) — на 1,0 (46,8±2,7) дптр, роговичного астигматизма — до 5,1±1,3 дптр (р≤0,05).
Через 6 мес КОЗ в среднем составляла 0,58±0,13, при этом отмечено ее повышение на 2 строки в 5 случаях, на 1 строку — в 9. Толщина роговицы в центре составила в среднем 442±89 мкм, Kave — 45,7±1,6 дптр, роговичный астигматизм — 4,8±1,5 дптр.
Через 1 год после лечения прослеживалась тенденция к улучшению средних показателей НКОЗ до 0,1±0,08 и КОЗ до 0,54±0,2, при этом положительная динамика зрения отмечена в 13 случаях, острота зрения не изменилась в 2 наблюдениях и снизилась — в 1 (р≤0,07). Преломляющая сила роговицы составила в среднем 45,9±4,9 дптр, роговичный астигматизм — 4,7±0,9 дптр.
Выявление ДЛ при ОКТ роговицы является одним из признаков успешного выполнения УФ-кросслинкинга. ДЛ обычно разделяет зоны интактной и подверженной УФ-кросслинкингу стромы и локализуется на глубине 50—80% толщины роговицы [24, 25]. Через 1 мес в 9 (56%) случаях ДЛ разной степени выраженности в виде узкой полосы разграничения между слоями стромы выявлялась в среднем на глубине 278±80 мкм (рис. 3). Через 3 мес только в 4 случаях прослеживалось наличие ДЛ, которая полностью исчезала к 6-му месяцу наблюдения. Через 12 мес специфических изменений роговицы не наблюдалось, сохранялось прежнее характерное локальное истончение в нижнем ее секторе.
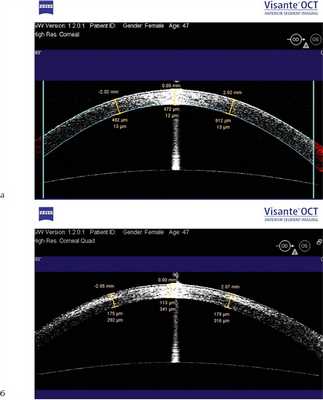
Рис. 3. Результат ОКТ роговицы высокого разрешения пациента до (а) и через 1 мес после (б) процедуры. Уплотнение роговицы и появление демаркационной линии.
Конфокальная микроскопия через 1 мес после процедуры показала незначительное снижение прозрачности экстрацеллюлярного матрикса, наличие «лакун» и появление «активных кератоцитов» — в средних слоях. В более глубоких слоях стромы отмечалось наличие линейных гиперрефлективных участков, а на уровне десцеметовой мембраны и эндотелиального слоя патологических изменений не выявлено. В сроки от 3 до 6 мес отмечались репопуляция кератоцитов в строме, уменьшение гиперрефлективности в глубоких слоях. К концу первого года исследования структура роговицы полностью восстанавливалась, что свидетельствовало об окончании репаративного процесса после проведения стандартного кросслинкинга.
ПМДР характеризуется медленным прогрессирующим течением. У пациентов, вошедших в наше исследование, ранние стадии заболевания протекали без значительных функциональных нарушений и ошибочно интерпретировались как миопия и сложный миопический астигматизм. Ввиду прогрессирующего течения эктазии коррекция аметропии ЖКЛ не всегда оказывалась удовлетворительной, отмечалась низкая толерантность к ЖКЛ.
По мнению многих исследователей, хирургические методы коррекции рефракционных нарушений — имплантация интрастромальных роговичных сегментов и колец, торических интраокулярных линз, появившихся в последнее десятилетие — не оказывают стабилизирующего влияния на течение кератэктатического процесса [26—30]. G. Wollensak и соавт. [15] методом электронной микроскопии подтвердили факт «склеивания» фибрилл и утолщение коллагеновых волокон роговицы под воздействием УФ-облучения в присутствии рибофлавина и как следствие — повышение биомеханических свойств. Внедрение нового метода лечения — кросслинкинга роговичного коллагена, оказывающего стабилизирующий эффект, расширило возможности поэтапной реабилитации пациентов с первичными кератэктазиями [20, 31—33]. Уменьшение радиуса кривизны роговицы и ее уплощение после кросслинкинга при кератэктазии описаны в ряде российских и зарубежных работ [15, 31—33]. S. Bayraktar и соавт. [7] указывали на рефракционную и функциональную эффективность кросслинкинга в виде монотерапии при ПМДР, при которой снижение преломляющей силы роговицы достигало 3,0 дптр и способствовало повышению остроты зрения. В ряде случаев кросслинкинг может способствовать повышению биомеханической резистентности роговицы, что обеспечивает стабилизацию течения процесса и «улучшает почву» для последующей хирургической коррекции аметропии [34].
Выявляемость ПМДР чаще в зрелом возрасте, отсутствие быстрого прогрессирования заболевания ограничивали применение кросслинкинга у данной категории пациентов. В литературе представлены единичные случаи применения кросслинкинга у больных с ПМДР, в том числе в сочетании с хирургическими методами коррекции аметропии. В частности, G. Kymionis и соавт. [33] показали эффективность и безопасность КР в сочетании ее с фоторефракционной кератэктомией. Сравнивая клиническое проявление двух эктазий и эффективность кросслинкинга при них, следует указать, что ПМДР реже имеет прогрессирующее течение, чем кератоконус, что и объясняет частое применение кросслинкинга при последнем. Динамика клинико-морфологических показателей после проведения КР при этих кератэктазиях сопоставима. Однако следует отметить, что в сравнении с ПМДР при кератоконусе рефракционный эффект КР выражен из-за более центрального расположения эктазии, равномерного уплощения ее, меньшего цилиндрического компонента рефракции. При ОКТ частота выявляемости ДЛ после КР реже при ПМДР, что требует дальнейшего изучения. По данным исследователей, конфокальная микроскопия роговицы показала меньшую диагностическую информативность при ПМДР в сравнении с кератоконусом. Это связано с локализацией зоны эктазии на периферии, что делает исследование не всегда возможным, а результаты достоверными [35]. Характерные структурные изменения роговицы после кросслинкинга при обоих видах эктазий схожи. Влияние процедуры на роговичный коллаген, эктрацеллюлярный матрикс, появление «активных кератоцитов» имеют одинаковую картину при двух видах первичных кератэктазий.
Проведенные собственные исследования показали, что роговичный кросслинкинг при ПМДР способствует изменению архитектоники роговицы и уплотнению ее стромы, равномерному уплощению и выравниванию центральной ее зоны, снижению роговичного астигматизма, что обеспечивает улучшение функциональных результатов и создает благоприятные условия для последующих рефракционных мероприятий: контактной коррекции, интрастромальной кератопластики (имплантация сегментов и колец). Выявлены снижение средней преломляющей силы роговицы более чем на 1,0 дптр, улучшение КОЗ на 1—2 строки. При анализе анатомо-функциональных результатов становится ясно, что повышение остроты зрения происходит за счет уменьшения астигматического компонента рефракции вследствие общего выравнивания и уплощения роговицы. Это позволяет рассматривать кросслинкинг как первый этап в системе реабилитации пациентов с прогрессирующей формой ПМДР.
Заключение
Пеллюцидная маргинальная дегенерация роговицы представляет собой разновидность первичных кератэктазий. Это, как правило, двусторонний, медленно прогрессирующий процесс с характерной клинической картиной и поздним началом. Проведение фотохимического кросслинкинга роговицы при прогрессирующем течении заболевания способствует улучшению оптометрических показателей и стабилизации процесса.
Семейная дегенерация роговицы, патология обмена кальция и глухота
Офтальмоплегия, дегенерация сетчатки, нарушение проводимости сердца и глухота
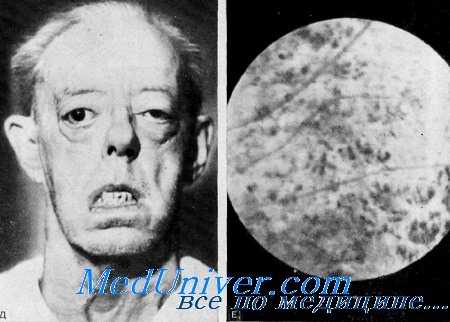
Kearns и Sayre впервые описали синдром «пигментного ретинита, наружной офтальмоплегии и полной сердечной блокады» у 2 неродственных больных. Kearns добавил соответственно еще 9 больных — 6 с «полным» и 3 с «неполным» синдромом, т. е. без кардиомиопатни. Дополнительные наблюдения были представлены Jager, Fred, Butler и Carries, Drachrnan, Danta с соавт, и Gadoth.
Клинические данные. Данные осмотра. Если синдром выявляется в раннем детстве, то у больных отмечается тенденция к низкому росту и слабому телосложению.
Орган зрения. Первые очевидные симптомы болезни, птоз и прогрессирующая наружная офтальмоплегия, у большинства больных появились во втором десятилетии жизни. Вскоре вслед за ними выявились пигментная дегенерация сетчатки и атрофия зрительных нервов. Хотя у некоторых больных главным расстройством являлся птоз, многие из них компенсировали его путем запрокидывания головы назад. Экзофтальм, обнаруженный у нескольких больных, возможно, является следствием слабости глазных мышц. Пигментная дегенерация сетчатки характеризуется рассеянным по всей сетчатке, иногда расположенным маленькими группами чистым пигментом. Вокруг диска зрительного нерва отмечаются чистые участки.
Типичные «костные тельца» не наблюдаются. Артерии сетчатки не сужены, но сама сетчатка истончена. Часто снижена острота зрения.
Нервная и мышечная система. Бульбарпые нарушения проявляются афонией, дисфагией и хриплым голосом. Голос слабый, речь с носовым оттенком, обусловленным неполным закрытием надгортанника. У большинства больных наблюдаются атрофии мышц лица, манифестирующиеся нарушениями открывания и смыкания век. В некоторых случаях была обнаружена мпопатия в проксимальных отделах плечевого и тазового пояса. У других больных отмечалась мышечная слабость и утрата чувствительности в дистальных отделах конечностей, что позволяло предположить периферическую нейропатию.
Мозжечковая атаксия, симптомы поражения кортико-спинального тракта (положительный симптом Бабинского, гиперрефлексия или арефлексия) и изменения психики по органическому типу являлись непостоянными симптомами.
Сердечно-сосудистая система. У большинства больных отмечался дефект сердечной проводимости. Блок одной из ножек пучка Гиса мог прогрессировать, приводя к атриовентрнкулярной блокаде различной степени выраженности. У ряда больных наблюдались синкопальные эпизоды типа Стокса — Адамса, связанные с изменением сердечного ритма.
Эндокринная система. У некоторых больных отмечалось недостаточное развитие половой сферы.
Орган слуха. У большинства больных была обнаружена потеря слуха смешанного типа, хотя у некоторых была установлена только нейросенсорная глухота. Потеря слуха была резче выражена па высоких частотах. Тест Бекеши и SISI-тест вызывали подкрепление феномена только на высоких частотах, но не на низких. Эти данные позволили предположить локализацию поражения в улитке или в стволе мозга
Вестибулярная система. Калорические пробы выявили значительное снижение реакции на стимуляцию вестибулярного аппарата или даже полное отсутствие реакции.
Лабораторные данные. Электроретипографпя показала ослабление реакции. Электроэнцефалографические изменения были неспецифическими и распределялись от грубой диффузной медленноволновой активности со спайками и комплексами волн в теменных и затылочных областях до легких медленных волн в лобных долях. Электромиография и данные биопсии позволили предположить миопатию.
У всех больных было повышено содержание белка в спинномозговой жидкости.
Содержание в сыворотке крови фнтаповой кислоты, SGOT, СРК и LDH не было повышенным. Не выявлено альфа-, бета-липопротеинемии, нарушения всасывания или акаптоцитоза. У некоторых больных была снижена экскреция стероидов.
Патология. Биопсия глазных мышц обнаружила увеличение эндомизиальной и перимизиальной соединительной ткани, разные размеры волокон и несколько центральное расположение ядер. Не было обнаружено увеличения мышечных волокон, сарколемных ядер или воспалительных инфильтратов.
Наследственность. Все случаи были спорадическими. Дальнейшее обсуждение см. ниже.
Диагноз. Прогрессирующая наружная офтальмоплегия с птозом или без него может встречаться как изолированная патология или быть обнаружена в сочетании с дюжиной или более синдромальных комбинаций, некоторые из которых могут оказаться ложными. Этому вопросу посвящен обзор Drachrnan. Синдром Рефсума должен быть исключен на основании данных о нормальном содержании фитановой кислоты в сыворотке крови.
Существует много сходных наблюдений, что затрудняет точную классификацию. Olson с соавт., DiMauro с сотр., а также Morgan-Hughes и Mair сообщили о больных с симптомами, описанными при данном синдроме. У них были обнаружены накопления гликогена и гигантские митохондрии с паракристаллическими включениями. Все случаи были спорадическими. Однако Taniura с сотр. описали 3 больных сибсов, родители которых состояли в кровном родстве. Возможно, мы имеем дело с этиологической гетерогенностью.
Лечение. Больные должны быть направлены к кардиологу для лечения дефекта сердечной проводимости. Птоз может быть исправлен при помощи пластической операции.
Прогноз. Различные симптомы синдрома прогрессируют, но в большинстве случаев заболевание не приводит к заметному укорочению жизни.
Выводы. Характеристика синдрома включает: 1) прогрессирующую наружную офтальмоплегию 2) пигментную дегенерацию сетчатки; 3) птоз; 4) атрофию зрительных нервов; 5) экзофтальм у некоторых больных; 6) бульбарные расстройства — дисфонию, дисфагию, хриплый голос; 7) проксимальные мышечные атрофии; 8) мозжечковую атаксию и иногда симптомы поражения кортнко-спинального тракта; 9) дефект сердечной проводимости; 10) недоразвитие половой сферы; 11) смешанную глухоту; 12) вестибулярную патологию.
Синдром Харбойяна: врожденная дистрофия роговицы и глухота
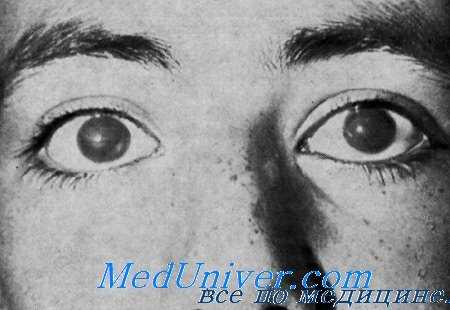
Комбинация врожденной дистрофии роговицы с прогрессирующей нейросенсорной глухотой описана у 2 из 10 епбеов, рожденных от одного двоюродного брака, и у 1 из 10 сибсов, рожденных от другого двоюродного брака того же самого отца (Harboyan et al.).
Орган зрения. С рождения отмечалась белесоватая мутность роговицы со снижением остроты зрения. Офтальмологическое исследование 3 больных в возрасте от 12 до 50 лет обнаружило такие же изменения. Эпителий роговицы был шероховатым, по не был окрашен. Строма ее была отечной, утолщенной и гомогенно белой. Острота зрения у 12-летнего мальчика на оба глаза была снижена до 20/200. У мужчин 28 и 50 лет счет пальцев был возможен только на расстоянии мене 1 м. У женщин 28 и 50 лет счет пальцев также был доступен только на расстоянии менее 1 м.
У молодых женщин было обнаружено повышение внутриглазного давления, тогда как у пожилых женщин, после наступления менопаузы, отмечалось ухудшение зрения. Эти наблюдения позволяют предположить некоторое прогрессирование нарушений зрения.
Орган слуха. Потеря слуха была впервые отмечена в возрасте от 10 до 25 лет. В дальнейшем она медленно прогресировала. На аудиограмме у 12-летнего мальчика была обнаружена двусторонняя нейросеисорная глухота в диапазоне от 20 до 50 дБ, выраженная более резко на высоких частотах. У 2 его полусибсов (сестер) потеря слуха достигала 40—70 дБ. Различение речи составляло 90—100%, SISI и tonedecay-тесты были отрицательными.
Вестибулярная система. У всех 3 больных калорические вестибулярные пробы были нормальными.
Лабораторные данные. Рутинные лабораторные исследования, включая определение уровня мукополисахаридов в моче, были нормальными.
Наследственность. Здоровые родители и существование кровного родства между ними ясно указывают на то, что синдром наследуется по аутосомно-рецессивному типу.
Диагноз. Врожденная дистрофия роговицы может быть изолированной патологией, наследующейся по аутосомно-рсцессивному типу (Маumenee). Помутнение роговицы может быть симптомом некоторых мукополисахаридозов (синдром Гурлер, Шейе и Марото— Лами). Эти заболевания, однако, клинически вполне различимы. При врожденной глаукоме встречаются помутнение роговицы, светобоязнь, расширение роговицы, увеличение внутриглазного давления и слезотечение. При синдроме Когана отмечаются светобоязнь и инъецирование глаз, а кератит расположен глубоко в строме.
При синдроме Когана отмечаются также шум в ушах, резкое головокружение и, наконец, выраженная глухота с утратой вестибулярной функции. Необходимо также исключить врожденный сифилитический кератит.
При аутосомно-рецессивной дистрофии роговицы Фера (Fehr) изменения роговицы становятся очевидными в течение первого десятилетия жизни, при настоящем же синдроме имеется врожденная дистрофия роговицы (Francois). Мы осведомлены только об одном случае сочетания дистрофии роговицы Фера с врожденной невральной глухотой. Больной был рожден от кровно-родственного брака (Моrо, Ameidi).
Другим возможным примером синдрома Харбойяна являются сибсы, описанные Scialfa с сотр. Родители были кровными родственниками. Однако у этих сибсов наблюдались умственная отсталость и клинодактилия V пальцев.
Лечение. Показаны трансплантация роговицы и лечение глаукомы. Могут быть использованы слуховые аппараты.
Прогноз. Потеря зрения и слуха медленно прогрессирует.
Выводы. Главные черты синдрома: 1) аутосомно-рецессивное наследование; 2) врожденная дистрофия роговицы с тенденцией к медленному прогрессированию; 3) выявляющаяся в детстве, медленно прогрессирующая нейросенсорная глухота.
Синдром Норри. Глазо-слухо-церебральная дегенерация
Синдром, характеризующийся помутнением хрусталика, атрофией радужной оболочки и пролиферацией ретролентальных масс, возможно, был впервые описан Fernandez-Santos, хотя может быть Clarke отметил его раньше.
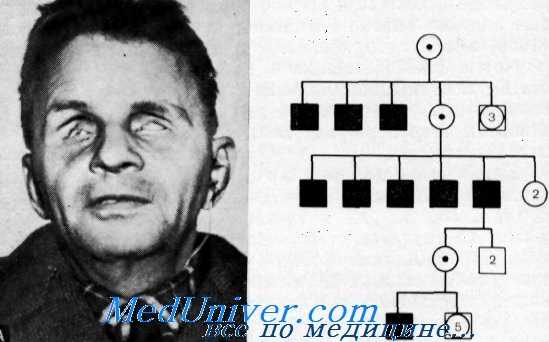
Norrie сообщил о двух семьях с этим заболеванием в своей статье, посвященной причинам слепоты у детей. Впоследствии было описано около 30 семей с этим синдромом (Brini et al.). Warburg провел обширное исследование этих и других семей в скандинавских странах.
Клинические данные. Орган зрения. С, возрастом глазные изменения драматически нарастают (Holmes). При рождении глаза нормальные, но уже в течение нескольких первых дней или недель жизни на сетчатке обоих глаз появляется желтый пигмент. К этому времени офтальмологическое исследование обнаруживает позади прозрачного хрусталика белые васкуляризированные массы. Fradkin нашел аниэокорию, мелкую переднюю камеру, ретролентальпую сосудистую пленку и геморрагию стекловидного тела. В течение дошкольного возраста развивается катаракта, роговица становится непрозрачной и глаза начинают сморщиваться. В школьном возрасте у большинства больных имеются изменения роговицы, хотя у некоторых очевидны атрофия радужной оболочки и синехии. К 10-летнему возрасту изменения глаз прекращаются.
Однако из-за болей в отдельных случаях приходится производить энуклеацию (Warburg).
Нервная система. Среди 30 больных, сведения о психическом состоянии которых имелись, около 1/3 были глубоко слабоумными, у 1/3 была легкая умственная отсталость и у 1/3 — нормальный интеллект. В более тяжелых случаях деградация начиналась на третьем или четвертом году жизни, в легких случаях распад психики происходил медленно. Некоторых больных в пятом — шестом десятилетии жизни необходимо было поместить в специальные учреждения (Forssman, Warburg, Lomickova, Raska).
Opган слуха. В исследовании Warburg в шести семьях у 11 из 35 больных был обнаружен дефект слуха. В нескольких случаях глухоту нельзя было установить из-за глубокого слабоумия. Потеря слуха прогрессивно нарастала во втором и третьем десятилетии жизни. У 6 из 13 больных, которым произвели оценку слуха, была обнаружена глухота от легкой до резко выраженной степени. Па аудиограммах обычно выявлялась симметричная нейросенсорная глухота в пределах 20—100 дБ.
Вестибулярная система. Warburg не производил вестибулярных проб, так как у нескольких больных был нистагм, а нескольким была произведена энуклеация.
Лабораторные данные. Электроэнцефалограммы у больных с глубоким слабоумием выявили выраженную диффузную патологию со спайками.
Хроматография аминокислот мочи и сыворотки крови, пммупофорез сыворотки крови и спинномозговой жидкости, а также содержание белка в спинномозговой жидкости были нормальными.
Патология. Исследование глаз показало, что полость стекловидного тела заполнена коллагеиовымн волокнами и васкулярнзпровапной рубцовой тканью. Кроме того, была обнаружена пролиферация пигментного эпителия сетчатки. Нижний слой сетчатки отсутствовал и был заменен недифференцированной глиальной тканью. Глазные яблоки обычно были маленькими, а роговица — куполообразной. Сосудистая оболочка была покрыта передней поверхностью радужной оболочки. Хорпопдеа была отечной и содержала застойные кровеносные сосуды. Отмечалось катарактное изменение хрусталика. Разрез зрительного нерва показал миелинизпрованиые волокна только па периферии, а в остальных частях нерва соединительную ткань (Warburg, Touenes, Roca).
Зрительный тракт был маленьким пли нитевидным и состоял главным образом, из глин, а латеральные коленчатые тела были почти в половину нормального размера с соответствующим уменьшением числа ганглиозных клеток. Поверхностные отделы мозга, включая медиальную поверхность затылочной доли мозга, были меньшего, чем в норме, размера. В коре мозжечка обнаружено несколько неправильное расположение нервных клеток с неполным образованием слоев.
Наследственность. Во многих опубликованных семьях синдром встречался в нескольких поколениях. Заболевание проявлялось только у мужчин. Все сыновья и дочери больных мужчин были здоровыми. Некоторые больные мужчины имели от дочерей больных внуков. Все приведенные родословные совместимы с Х-сцепленным наследованием. У женщин-носительниц не обнаружено дефектов зрения или слуха. Гомозиготные женщины, возможно, были описаны Кларком (Clarke). Изучение сцепления было безуспешным (Nance et al.,).
Диагноз. У нескольких больных с синдромом Норри была ошибочно диагностирована ретинобластома. Это заболевание должно быть исключено на основании другой глазной патологии, которая наблюдается при синдроме Норри. Больные с ювенильной колобомой сетчатки имеют значительно лучшее зрение, чем больные с синдромом Норри. Необходимо также исключить серповидную сетчатку и отслойку сетчатки, токсоплазмоз, рстролептальную фиброплазую, травмы с массивным фиброзом сетчатки. Может потребоваться дифференциация с метастатической офтальмией (почти всегда односторонней), с Х-сцепленной врожденной катарактой, а также с офтальмией новорожденных и X-сцепленной микрофтальмией (Hansen).
Лечение. Наблюдающаяся в отдельных случаях боль в глазах может быть облегчена посредством энуклеации. При потере слуха могут быть эффективными слуховые аппараты, если их использованию не препятствует глубокая умственная отсталость.
Прогноз. Все больные слепнут. Нейросенсорная глухота от умеренной до резко выраженной степени не выглядит прогрессирующей. Слабоумие же прогрессирует. Больные дети только на первом или втором году жизни производят впечатление интеллектуально полноценных. Некоторые больные умирают в психиатрических больницах при явлениях острого психоза.
Выводы. Главная характеристика синдрома Норри включает: 1) Х-сцеплснное рецессивное исследование: 2) изменения со стороны глаз, заключающиеся в глиальной пролиферации сетчатки, катаракте и микрофтальмии; 3) слабоумие от легкой до глубокой степени примерно в 2/3 случаев; 4) нейросенсорную глухоту от легкой до резко выраженной степени примерно у 1/3 больных.
1. Эндотелиальная дистрофия роговицы Фукса — К1, К2, К3.
2. Задняя полиморфная дистрофия роговицы — К1 или К2.
3. Врожденная эндотелиальная дистрофия — К1.
4. X-связанная эндотелиальная дистрофия роговицы — К2.
Далее приводятся данные о наиболее распространенных в офтальмологической практике и клинически значимых дистрофиях роговицы.
Дистрофия эпителиальной базальной мембраны (ДЭБМ) — одна из часто встречающихся дистрофий. Хотя она включена в представленную классификацию и поражает один слой роговицы, что типично для дистрофических поражений, только небольшое число случаев имеет доказанное наследование — ген TGFBI (transforming growth factor beta-induced), локус 5q31 [7, 11]. В большинстве наблюдений состояние считают специфической реакцией роговицы на внешние воздействия, т. е. дегенерацией, которая обусловлена аномальным восстановлением, созреванием и формированием базальной мембраны, приводящим к образованию патологических комплексов адгезии и ослабленному прикреплению эпителия к строме [7, 12]. Дистрофия встречается у 2% в популяции, чаще у женщин. При биомикроскопии в эпителиальном слое роговицы выявляются сероватые включения, тонкие линии, микрокисты. Эти образования формируют различные типы рисунка в субэпителиальном слое — точечные и микрокистные изменения, географический рисунок, рисунок «отпечатка пальца», «булыжной мостовой» (рис. 1). Рис. 1. Дистрофия эпителиальной базальной мембраны. Со временем они могут менять свою локализацию и тип рисунка [13, 14]. Так как эти интраэпителиальные поражения полупрозрачны и могут быть очень маленькими по размеру, биомикроскопия должна проводиться с особой тщательностью с использованием различной техники освещения. При гистологическом исследовании выявляются интраэпителиальные разрастания или многослойные образования базальной мембраны эпителия, которые формируют географический рисунок или рисунок «отпечатка пальца». Точечные изменения представляют собой псевдокисты, содержащие твердые частицы в цитоплазме и скопление клеточного детрита между патологическими разрастаниями мембраны либо в слоях эпителия [5—8]. Симптоматика дистрофии тесно связана с синдромом рецидивирующей эрозии роговицы и временным незначительным снижением остроты зрения, что чаще имеет место у пациентов старше 30 лет. Считается, что от 10 до 30% пациентов с ДЭБМ подвержены рецидивам эрозии роговицы и у 50% больных с рецидивирующей эрозией роговицы выявляются признаки ДЭБМ. При изменениях базальной мембраны в оптическом центре возможен неправильный астигматизм. В большинстве случаев дистрофия протекает бессимптомно [6].
Дистрофия Рейс—Буклера — аутосомно-доминантная (ген TGFBI, локус 5q31), билатеральная, центральная дистрофия роговицы с первичным поражением боуменового слоя, начальные признаки которой появляются в первые годы жизни в виде поверхностных линейных и кольцевидных сероватых помутнений, сопровождающихся рецидивирующими эрозиями с выраженным болевым компонентом. К 20—30 годам помутнения прогрессируют, сливаются между собой, сохраняя характерный географический рисунок, распространяясь на среднюю периферию и глубже в строму, развивается диффузное стромальное неинтенсивное помутнение (хейз) [15, 16]. Частота эрозий с возрастом заметно уменьшается, ухудшается острота зрения из-за прогрессирования помутнений и неправильного астигматизма. Снижение остроты зрения при этой дистрофии более выражено, чем при сходной дистрофии Тиля—Бенке, из-за большего нарушения регулярности передней поверхности роговой оболочки глаза [7, 17]. При гистологическом исследовании видно, что эпителиальный слой теряет регулярную архитектонику, местами утолщен, повторяет измененный рельеф подлежащей соединительной ткани. Местами отсутствует базальная мембрана эпителия. Боуменов слой замещается листовидной фиброцеллюлярной соединительной тканью с зернистыми отложениями, которая в развитой стадии заболевания распространяется в передние слои стромы [7, 17, 18].
Для стромальных и ряда эпителиально-стромальных дистрофий характерным является отложение патологических веществ между коллагеновыми фибриллами или в кератоцитах. Это могут быть как нормальные метаболиты, представленные в избыточном количестве, как гликозамингликаны при макулярной дистрофии, так и вещества, не встречающиеся в роговой оболочке в норме (амилоид, холестерол, гиалин) [8, 17].
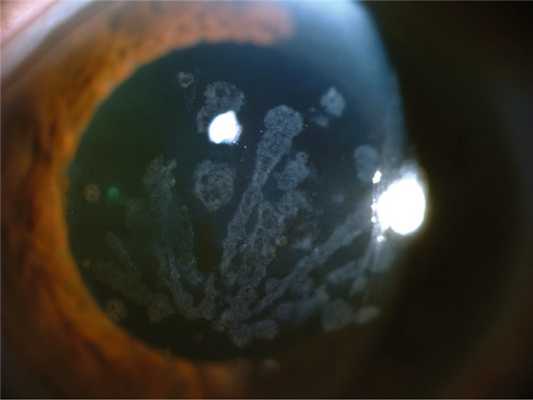
Зернистая дистрофия роговицы, тип 1 (классическая) имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31), билатеральная, симметричная. Заболевание проявляется в первую декаду жизни. Осмотр за щелевой лампой выявляет хорошо видимые гранулы, которые кажутся белыми при прямом освещении. При ретроиллюминации эти гранулы состоят из чрезвычайно мелких, полупрозрачных точек, выглядят как вакуоли, стеклянные осколки или измельченные крошки. Помутнения не захватывают лимбальную зону (рис. 2). Рис. 2. Зернистая дистрофия роговицы, тип 1. У детей коричневые гранулы, образующие вихревидный рисунок, расположены относительно поверхностно и распространяются к слою Боумена. В дальнейшем гранулы простираются в глубокие слои стромы вплоть до десцеметовой мембраны [6, 17]. Блики и светобоязнь являются относительно ранними симптомами. Так как на начальных этапах регулярность передней поверхности не нарушена, острота зрения остается высокой. С возрастом помутнения прогрессируют, сливаются между собой, острота зрения постепенно снижается. Иногда имеют место рецидивирующие эрозии. В развитых стадиях острота зрения редко снижается — менее 0,1. Как правило, к 50—60 годам необходима кератопластика, после которой вероятны рецидивы в течение нескольких лет (от 1 года до 20 лет) [7, 8, 18].
Зернистая дистрофия роговицы, тип 2, считается комбинированной — зернисто-решетчатой. Также часто именуется дистрофией роговицы Авеллино по местности, где проживала семья, страдавшая таким заболеванием. Имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31). По данным световой микроскопии помутнения роговицы простираются от базального эпителия до глубокой стромы. Помимо типичных для зернистой дистрофии 1-го типа гиалиновых отложений, в строме присутствует амилоид [5]. Заболевание начинается в первой декаде жизни, но выявляется чаще в подростковом возрасте или в ранней зрелости. Начальными признаками дистрофии, которые выявляются под щелевой лампой, считаются поверхностные стромальные крошечные белесые точки. На следующем этапе между поверхностной и средней стромой появляются кольцевидные или в форме снежинки помутнения. У некоторых пациентов в более глубоких слоях также имеют место решетчатые линии. Как правило, эти линии расположены глубже, чем стромальные помутнения в виде снежинок. На заключительном этапе полупрозрачные плоские крошковидные помутнения, расположенные поверхностно, могут сливаться между собой. У некоторых пациентов проявления дистрофии ограничиваются только множеством белых точек. Пациенты с зернистой дистрофией 2-го типа имеют меньше помутнений, чем лица с зернистой дистрофией 1-го типа. Острота зрения уменьшается с возрастом, так как постепенно вовлекается центральная зона. При эрозиях, которые, как правило, не имеют тяжелого течения, возникают болевые ощущения [6, 17, 19]. Заболевание прогрессирует медленно, несколько быстрее при гомозиготной форме.
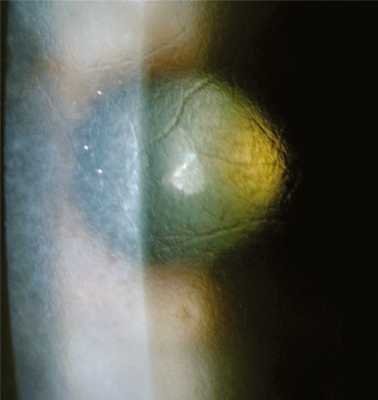
Решетчатая дистрофия роговицы, тип 1, считается классической решетчатой дистрофией роговицы, имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31). При гистологическом исследовании выявляются отложения амилоидного вещества в строме, которые нарушают ламеллярную архитектонику роговицы. Также могут отмечаться эпителиальная атрофия и дегенеративные нарушения базальных эпителиальных клеток, очаговое истончение или отсутствие слоя Боумена [5]. При биомикроскопии уже к концу первого десятилетия жизни видны тонкие ветвящиеся, отражающие свет линии и/или субэпителиальные беловатые овальные точки. Линии появляются в центре достаточно поверхностно, распространяясь на периферию и в глубь роговой оболочки глаза, но оставляя периферию, десцеметову мембрану и эндотелий интактными (рис. 3). Рис. 3. Решетчатая дистрофия роговицы, тип 1. Диффузное стромальное помутнение матового цвета, как правило, развивается позже, сопровождается рецидивирующими эрозиями [7]. Дискомфорт, боль и зрительные нарушения иногда начинаются уже в первое десятилетие жизни. Зрительные нарушения становятся значительными к четвертому десятилетию, что часто является показанием к кератопластике в этом возрасте [6, 8, 20].
Решетчатая дистрофия роговицы, тип 2, была внесена в первую редакцию современной классификации лишь условно, исключена из второй, не считается дистрофией роговицы как таковой, а является глазным проявлением семейного амилоидоза. Сопровождается краниальной нейропатией, проявляющейся как лицевой парез, бульбарный паралич, периферической полинейропатией. При биомикроскопии изменения сходны с таковыми при типе 1. Чувствительность роговицы снижена или отсутствует. Острота зрения сохраняется достаточно высокой длительное время, так как дистрофия прогрессирует от периферии к центру роговицы. Развивается симптоматика синдрома сухого глаза. В пожилом возрасте могут возникать рецидивирующие эрозии роговицы. Течение заболевания медленно прогрессирующее. У большинства может не быть серьезных нарушений до седьмого десятилетия. Существует повышенный риск открытоугольной глаукомы [6, 7].
Макулярная (пятнистая) дистрофия роговицы. Имеет аутосомно-рецессивный тип наследования, ген CHST6 (сarbohydrate sulfotransferase 6 gene), локус 16q22.
При задней полиморфной дистрофии (ЗПД) тип наследования аутосомно-доминантный, выявлены различные локусы: для ЗПД1 — 20p11.2-q11.2; ЗПД2 — 1p34.3-p32.3; ЗПД3 — 10p11.22. Ген для ЗПД1 не выделен, для ЗПД2 — COL8A2, ЗПД3 — ZEB1. Морфологически на задней поверхности десцеметовой мембраны выявляются патологические слои коллагена с локальными веретенообразными или узелковыми разрастаниями. Среди эндотелия определяются участки многослойных эпителиоподобных клеток с микроворсинками и десмосомами [8, 24]. Заболевание проявляется часто асимметричными поражениями различной формы в глубоких слоях роговицы, включая узелковые, везикулезные (изолированные, в группах или сливные) и блистероподобные. Помутнения могут иметь вид «железнодорожных путей» и очаговых серых участков на уровне десцеметовой мембраны. Стромальный и эпителиальный отек из-за эндотелиальной декомпенсации развивается редко. Изменения в эндотелии часто не прогрессируют годами. Возможны медленное прогрессирование полиморфных везикул и утолщение десцеметовой мембраны на протяжении многих лет. Периферические иридокорнеальные сращения встречаются в 25% случаев. Примерно в 15% наблюдений уровень внутриглазного давления повышен [26]. Эндотелиальные изменения часто протекают бессимптомно. Редко имеется значительное прогрессирующие снижение остроты зрения в результате стромальных помутнений [6, 24, 27].
Лечение. Эффективной патогенетической терапии на современном этапе развития медицины не существует. Симптоматическое местное лечение направлено на купирование нарушений передней поверхности роговицы и уменьшение ее отека при тех дистрофиях, где он имеет место. Для этого применяются любриканты, препараты, способствующие регенерации, мягкие контактные линзы, гипертонические растворы [12]. Перспективным представляется использование современных инновационных слезозаменителей, одним из представителей которых является катионорм (Santen), созданный по технологии Novasorb на основе катионной эмульсии. Препарат эффективно восстанавливает слезную пленку, во многом воспроизводит физиологию естественной слезы, не содержит консервантов, что позволяет применять его длительное время без побочных эффектов.
Хирургическое лечение может включать абразивные вмешательства на передней поверхности роговицы для улучшения адгезии эпителиального слоя к глубжележащим слоям и отчасти для уменьшения ее иррегулярности. ФТК является эффективным методом при удалении помутнений передних слоев роговой оболочки на фоне поверхностных и некоторых видов стромальных дистрофий. Кроме того, при выполнении ФТК удается устранить иррегулярный астигматизм и улучшить адгезию эпителия. При рецидивах заболевания процедуру можно повторять несколько раз. При глубоких помутнениях, существенно снижающих остроту зрения, методом выбора является кератопластика. При относительно изолированном поражении слоев роговицы предпочтительными являются модификации послойной кератопластики (передняя послойная, глубокая послойная, эндотелиальная кератопластика). При одновременном вовлечении стромального и эндотелиального слоев целесообразно проводить сквозную кератопластику или ее модификации со сложным профилем операционного разреза. Как и после ФТК, после кератопластики вероятны рецидивы в сроки от 1 года до 20 лет, частота которых и степень снижения зрения зависят от вида дистрофии и могут варьировать индивидуально [8, 28, 29].
Дистрофии роговицы — группа медленно прогрессирующих невоспалительных поражений роговицы, большинство из которых отличаются вариабельностью фенотипических проявлений. Описанная в статье новая классификация сочетает в себе аспекты традиционного определения дистрофий роговицы с новыми генетическими, морфологическими и клиническими данными. Важно помнить о существовании подобной патологии и своевременно дифференцировать с острыми воспалительными процессами различной этиологии, которые требуют ургентной терапии. При дистрофиях целесообразное консервативное лечение, связанное, как правило, с нарушениями передней поверхности роговицы, является симптоматическим (любриканты, препараты, способствующие эпителизации, мягкие контактные линзы). Эффективной терапии этиопатогенетической направленности не существует. При значительном снижении остроты зрения возможно выполнение ФТК и различных модификаций кератопластики в зависимости от глубины поражения.
Читайте также:
- Механизмы переноса жирных кислот через плаценту
- Лечение менингококковой инфекции. Профилактика менингококковой инфекции. Менингококковая вакцина.
- Показания и подготовка к артроскопической аллотрансплантации мениска коленного сустава
- Порфирии - причины, классификация
- Рентгенограмма, КТ, МРТ при нетравматическом остеите лобковой кости