Сидеробластические анемии - причины, классификация
Добавил пользователь Евгений Кузнецов Обновлено: 24.01.2026
Сидеробластические анемии - причины, классификация
Сидеробластические анемии составляют особую группу заболеваний весьма разнообразной этиологии, которых, однако, объединяет общий знаменатель — наличие кольчатых сидеробластов в костном мозге (Bjorkman, Bowman). Последние рассматриваются патологическими и отличаются от нормальных — зерна феритина которых распространены по всей цитоплазме — тем, что железо располагается в гребешках митохондриев в виде железистых мицелиев (пластинок или пыли).
К другим общим характеристикам этих анемий можно отнести неэффективность эритропоэза, рост показателя железа в плазме и тканях и развитие гемохроматоза (Farid, Мунтяну и Попеску).
В медицинской литературе уже давно говорилось об этой болезни, включенной в крупную группу так называемых устойчивых анемий, поскольку они не поддавались никакому терапевтическому методу. Но постепенно, по мере освоения новых клинических и разведочно-лабораторных достижений, определилось, что некоторые из этих заболеваний отличаются четкой этиологией (отравление свинцом, длительное лечение изониазидом, ревматоидный артрит и пр.).
Другие — благоприятно реагируют на пиридоксинотерапию крупными дозами (сохраняя вместе е тем нормальный уровень пиридоксина в крови), в связи с чем получили название «анемий, реагирующих на пиридоксин». И наконец, наблюдение отдельных семейных случаев (Gooley, Bundles и Falls) сделало возможным отождествление наследственных форм болезни.
Однако имеется большая группа подобных заболеваний, с хорошо установленными клиническими и морфологическими характеристиками, получившая название первичной генуинной сидеробластической анемии, в отношение которой этиология еще не определена. Современная классификация этой болезни приведена ниже. Связанное с морфологическим пороком наличия кольчатых сидеробластов, это название было присвоено недавно (Bjorkman). Гейльмайер, основательно и многосторонне исследовавший эту болезнь, назвал ее сидероаккрестической анемией (вызываемой неиспользованием железа).

Классификация сидеробластических анемий
A. Первичные формы сидеробластических анемий:
I. Первичная приобретенная генуииная сидеробластическая анемия
II. Наследственная сидеробластическая анемия:
а) вид, связанный с полом, развивающийся за счет недостатка аминолевулин-синтстазы и копропорфириногеноксидазы (I тип по Гейльмейеру);
б) аутосомальный вид (поражающий лиц обоего пола) (II тип по Гейльмейеру),
Б. Анемия, поддающаяся пиридоксинотерапии
B. Вторичные формы сидеробластических анемий
I. Вызываемые токсическими веществами или медикаментами, в том числе, свинцом, гидразидом изоникотиновой кислоты, циклосерином, спиртом и пр.
II. Сопутствующие иным заболеваниям, в том числе, воспалительным (ревматизм, коллагеноз, заражение почек), крови (миелофиброз, истинная полицитемия, лейкемия, гемолитическая анемия, злокачественная анемия и пр.), прочим болезням (поздняя порфирия кожи, резецированный желудок, миксэдема и др.).
Общая гематологическая характеристика сидеробластических анемий:
1) Гипохромия или/и эритроцитный диморфизм.
2) Ретикулоцитопения.
3) Эритробластоз — костномозговой мегалобластоз.
4) Кольцевые сидеробласты.
5) Неполноценное кроветворение: внутрикостномозговое расплавление крови при высоких показателях косвенной гипербилирубинемии и уробилиногена в фекалиях, гиперсидеремия, клиренс и интенсивность и скорость метаболизма, высокий показатель Fe59, слабое-включение Fe59 в эритроциты, депонирование железа в запасах.
6) Нормальная продолжительность жизни эритроцитов.
7) Отсутствуют аномальные гемоглобины.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Наследственные сидеробластные анемии — разнородная по спектру генетических нарушений и выраженности клинических проявлений группа заболеваний.
Варианты наследования и патогенез
Наследственные сидеробластные анемии, сцепленные с Х-хромосомой. У большинства детей с сидеробластной анемией механизм наследования связан с Х-хромосомой. В связи с этим анемия диагностируется преимущественно у мальчиков и их близких родственников мужского пола по материнской линии (родные дяди и двоюродные братья). Редко имеется другой вариант наследования, не связанный с Х-хромосомой. В некоторых семьях заболевание возникает только у девочек, так как у мальчиков-гомозигот патология не совместима с жизнью.
В основе патогенеза лежит дефект синтеза 5-аминолевулинатсинтетазы (АЛК), участвующей в синтезе гема. Для нормальной работы этого фермента необходимо достаточное количество пиридоксина — витамина В6 (этим и объясняется эффективность лечения витамином В6). Активность фермента 5-аминолевулинатсинтетазы (АЛК-синтетазы) кодирует ген ALAS2, расположенный на Х-хромосоме. У больных сидеробластной анемией описано несколько различных мутаций данного гена.
Наследственные сидеробластные анемии с аутосомным типом наследования. Этот тип наследования встречается значительно реже, чем сцепленный с Х-хромосомой. Известны случаи как аутосомно-доминантного, так и аутосомно-рецессивного наследования. Мутации гена ALAS2 не определяются, поэтому терапевтический эффект пиридоксина отсутствует.
Спорадическая врожденная сидеробластная анемия. В мире описано около 20 случаев сидеробластной анемии, выявленной сразу после рождения, без признаков заболевания у других членов семьи. Вероятно, при этом имелись либо аутосомно-рецессивный тип наследования, либо появление новых мутаций гена ALAS2 в родительских половых клетках.
Митохондриальная цитопатия (синдром Пирсона). Синдром Пирсона — врожденное заболевание, которое обусловлено делециями или другими генетическими перестройками митохондриальной ДНК и клинически проявляется множественными органными поражениями. Одним из ранних признаков заболевания является тяжелая анемия, ассоциированная с наличием кольцевых сидеробластов в костном мозге. Продолжительность жизни детей с синдромом Пирсона обычно не превышает 2-3 года.
Синдром DIOMOAD. В основе большинства клинических проявлений заболевания, наследующегося по аутосомно-рецессивному типу, лежат дегенеративные процессы в нервной ткани, обусловленные, вероятно, наследственными дефектами метаболизма тиамина. Гематологическим проявлением синдрома является нормоцитарная сидеробластная анемия средней степени тяжести в сочетании с нейтропенией и выраженной тромбоцитопенией. Отмечен терапевтический эффект применения тиамина.
Классификация сидеробластных анемий
Данные клинических и лабораторных исследований при наследственных сидеробластных анемий
В большинстве случаев клиническая картина сидеробластной анемии, сцепленной с Х-хромосомой, не отличается от таковой при анемии с аутосомным типом наследования или других врожденных форм. Тяжелая анемия, как правило, диагностируется в младенчестве или раннем детстве. При менее выраженных проявлениях анемического синдрома или бессимптомном течении заболевание выявляется обычно у взрослых или даже пожилых больных.
Наряду с анемическим синдромом у всех больных определяются признаки избытка железа в организме, которые получили специальное название: синдром эритропоэтигеского гемохроматоза. Проявления этого синдрома достаточно разнообразны. Чаще всего определяются умеренная гепатомегалия и спленомегалия. Функция печени в большинстве случаев нарушена незначительно или не страдает вообще. При биопсии печеночной ткани определяются депозиты железа в гепатоцитах. У некоторых больных старше 30-40 лет при гистологическом исследовании находят признаки микронодулярного цирроза печени, протекающего, как правило, доброкачественно.
На фоне гемохроматоза поджелудочной железы могут определяться сахарный диабет или нарушения толерантности к глюкозе. Редко при объективном обследовании выявляется пигментация кожных покровов. Наиболее опасными проявлениями синдрома эритро-поэтического гемохроматоза являются тяжелые нарушения сердечного ритма и сердечная недостаточность, которые развиваются, как правило, на поздних этапах заболевания. В тяжелых случаях у детей может наблюдаться задержка роста и развития.
В анализе крови определяется анемия различной степени тяжести. При тяжелой анемии обычно имеются гипохромия, микроцитоз, анизо- и пойкилоцитоз, реже мишеневидные клетки и единичные сидероциты. При менее тяжелых формах анемии в мазке могут выявляться две популяции клеток: гипохромные микроциты и нормальные эритроциты. На гистограмме эритроцитов при этом образуется двухфазная кривая, отражающая различия в размерах эритроцитов. Уровень лейкоцитов и тромбоцитов обычно в норме, но при развитии гиперспленизма может снижаться. Количество ретикулоцитов в большинстве случаев в норме или незначительно повышено.
В миелограмме выявляется гиперплазия эритроидного ростка на фоне нормобластического типа кроветворения и повышенное количество сидеробластов. В редких случаях (при сопутствующем дефиците фолиевой кислоты) может определяться мегалобластический тип кроветворения.
При биохимическом исследовании обычно выявляется незначительная гипербилирубинемия, повышение уровня ферритина и снижение уровня трансферрина, и увеличение его сатурации. При анемии, сцепленной с Х-хромосомой, имеется снижение активности АЛК-синтетазы.
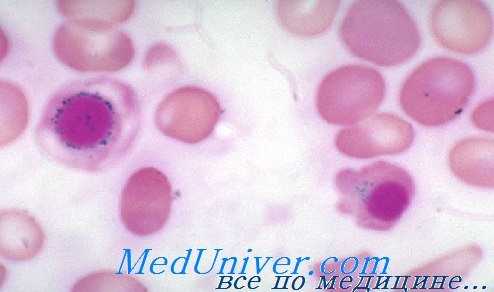
Мазок при сидеробластной анемии
У больных с синдромом Пирсона диагностируется рефрактерная сидеробластная анемия, которая сочетается с признаками экзокринной недостаточности поджелудочной железы, эпизодами молочнокислого ацидоза и прогрессирующей почечной и печеночной недостаточностью. Анемия выявляется, как правило, после рождения и носит нормоцитарныи или макроцитарный характер. Уровень ретикулоцитов чаще всего снижен. В большинстве случаев имеются нейтропения и тромбоцитопения различной степени тяжести. При электрофорезе гемоглобина обычно определяется повышение уровня Hb F. Костный мозг гиперклеточный или нормоклеточный, выявляются кольцевые сидеробласты.
Лечение и прогноз наследственных сидеробластных анемий
Всем больным наследственными сидеробластными анемиями необходимо начинать терапию пиридоксином (витамин В6), которая эффективна в среднем в 1/3 случаев. Доза витамина В6 обычно составляет 50-100 мг в сутки. Выраженность терапевтического эффекта у пациентов, ответивших на лечение, различна. В большинстве случаев появляется ретикулоцитоз и уровень гемоглобина в течение 1-2 месяцев лечения постепенно повышается до нормы или субнормальных значений. Морфологические изменения эритроцитов при этом сохраняются.
При отсутствии оптимального эффекта на фоне терапии пиридоксином уровень гемоглобина стабилизируется, но не достигает нормальных значений. Необходимо проведение поддерживающей терапии витамином В6, при отсутствие которой концентрация гемоглобина через несколько месяцев снижается до исходных величин. При выявлении мегалобластического типа кроветворения показано лечение фолиевой кислотой. Пациентам с тяжелой анемией, у которых отсутствует эффект витамина В6, по показаниям проводятся периодические трансфузии эритроцитарной массы. Это уменьшает выраженность анемии и предотвращает задержку роста и развития у детей.
Для профилактики гемосидероза показана терапия дефероксамином, в которой нуждаются прежде всего больные с тяжелой анемией, получающие гемотрансфузионную терапию. Предпочтительно проведение 12-часовых подкожных инфузий препарата в дозе 40 мг/(кг-день) каждые 5 дней недели (этот режим обладает минимальной токсичностью). Спленэктомия при наследственных формах сидеробластной анемии довольно часто осложняется тромбоэмболией, в ряде случаев с летальным исходом, поэтому оперативное лечение используется редко.
Сидеробластная анемия ( Сидероахрестическая анемия )
Сидеробластная анемия – это приобретенное или наследственное заболевание крови, характеризующееся гипохромией эритроцитов, высоким уровнем сывороточного железа с отложением в тканях органов. Клиническая картина представлена анемическим синдромом (головокружение, утомляемость, бледность кожи), гемосидерозом внутренних органов (гепатомегалия, кардиомегалия, ХПН), изменением картины крови. Диагноз выставляется на основании клинических и биохимических анализов (ОАК, сывороточное Fe, ферритин, трансферрин, витамин В6), генодиагностики, исследования миелограммы. Лечение может включать гемотрансфузии, витамино-, гормонотерапию, хелаторную терапию.
МКБ-10

Общие сведения
Сидеробластные (сидероахрестические) анемии включают несколько гетерогенных синдромов, которые протекают с нарушением синтеза гема, несмотря на повышенное содержание железа в плазме. Популяционная частота сидеробластных анемий (СБА) не выяснена ввиду их клинической неоднородности, однако известно, что приобретенные формы встречаются чаще наследственных. Врожденные анемии обычно манифестируют в детстве, но могут оставаться нераспознанными вплоть до зрелого возраста. Первичная приобретенная сидеробластная анемия диагностируется преимущественно у лиц среднего и пожилого возраста.
Причины
Врожденная сидеробластная анемия
Врожденные СБА связаны с мутациями в генетическом материале. Они различаются по типу наследования и патогенетическому механизму:
- Х-сцепленные сидеробластные анемии. Один вариант обусловлен дефектами гена ALAS2 ‒ синтазы дельта-аминолевуленовой кислоты II типа (локус Хр11.21). Другой тип болезни связан с трансгенациями hABC7 (Xp13.1-q13.3), кодирующего белок, участвующий в транспорте гема.
- Аутосомно-рецессивная сидеробластная анемия. Генетически гетерогенная группа. Может возникать при дефекте гена WFS1 (4p16.1), отвечающего за экспрессию трансмембранного белка вольфрамина. В этом случае СБА диагностируется в рамках синдрома Вольфрама (DIDMOAD). Другая форма развивается при дефектах в гене SLC25A38, кодирующем белок-транспортер глицина, необходимый для синтеза гема.
- Митохондриальная цитопатия. Вызвана делециями митохондриальной ДНК, клинически проявляется развитием синдрома Пирсона.
- Спорадические сидеробластные анемии. Обусловлены появлением вновь возникших генетических мутаций, которые отсутствуют у родителей и других членов семьи.
Приобретенная сидеробластная анемия
Идиопатические (первичные) СБА ‒ рефрактерные анемии с кольцевыми сидеробластами ‒ рассматриваются как разновидность миелодиспластического синдрома (МДС) и занимают в его структуре 5-15%. Чаще возникают как следствие лучевой или цитостатической терапии, проводимой по поводу коллагенозов, гемобластозов, различных онкопроцессов. Риск развития МДС повышен у курильщиков, лиц, контактирующих с химическими веществами (бензином, растворителями, инсектицидами).
Вторичная сидеробластная анемия может быть этиологически связана со следующими факторами:
- прием антибактериальных и туберкулостатических препаратов (хлорамфеникол, линезолид, изониазид, циклосерин);
- интоксикация свинцом;
- хронический алкоголизм;
- нарушение баланса витаминов и микроэлементов: дефицит пиридоксина (витамина В6), недостаток меди, избыток цинка;
- коллагенозы;
- болезни крови (полицитемия, гемолитическая анемия).
Патогенез
Независимо от многообразия причин все они тем или иным путем приводят к нарушению образования гема. Гем является соединением, состоящим из протопорфирина и атома железа (Fe2+). При его связывании с белком глобином образуется гемсодержащий хромопротеин гемоглобин, главная функция которого заключается в транспорте кислорода к тканям, а углекислоты из тканей.
При СБА в результате дефектов ферментных систем, дефицита пиридоксальфосфата, токсического влияния или других факторов нарушается биосинтез протопорфирина, что делает образование гема невозможным, несмотря на достаточный уровень Fe в сыворотке крови. Развивается гипохромная анемия. В условиях, когда железо не используется клетками, оно начинает накапливаться в тканях и внутренних органах с развитием их дисфункции.
Классификация
Сидеробластные анемии делятся на 2 большие группы – наследственные и приобретенные, внутри которых выделяют конкретные клинические формы, связанные с определенными этиофакторами:
1. Наследственные сидеробластные анемии:
- Х-сцепленные;
- аутосомные (могут быть пиридоксин-рефрактерными и пиридоксин-зависимыми);
- митохондриальные болезни.
2. Приобретенные сидеробластные анемии:
- первичные ‒ клональные СБА в рамках миелодиспластического синдрома;
- вторичные ‒ обратимые СБА (лекарственные, токсические, алиментарные);
- на фоне других заболеваний (системных, гематологических и пр.)
Симптомы сидеробластной анемии
В клинической картине сидероахрестических анемий преобладают две группы симптомов: циркуляторно-гипоксические и недостаточность органов, обусловленная гемосидерозом. Гематологические изменения выявляются при диагностическом исследовании крови и костного мозга.
Гипоксический синдром сопровождается недомоганием и слабостью, тахикардией, снижением АД. Характерны жалобы на головокружение, ноющие боли в сердце, появление «мушек» перед глазами, одышку при движениях. Отмечается сухость и бледность кожи, ломкость ногтей. У детей ухудшается запоминание учебного материала, снижается успеваемость.
Депонирование железа приводит к развитию гепатоспленомегалии, а в поздних стадиях – циррозу печени. Поражение поджелудочной железы сопровождается манифестацией сахарного диабета. Гемосидероз миокарда проявляется кардиомегалией, нарушением ритма, сердечной недостаточностью. Отложение железа в тканях мужских половых желез вызывает вторичный гипогонадизм. При поражении легких возникает дыхательная недостаточность, почек – ХПН.
Осложнения
Перегрузка железом вызывает необратимые органные поражения, приводящие к стойкой функциональной недостаточности сердца, печени, почек, эндокринных органов. Таким пациентам требуется пожизненная заместительная и поддерживающая терапия, у них снижена общая продолжительность жизни. Течение идиопатической формы сидеробластной анемии у 5-10% пациентов осложняется острым лейкозом, рефрактерным к полихимиотерапии.
Диагностика
Основанием для выставления диагноза «сидеробластная анемия» служат клинические и лабораторные данные. Больных консультирует врач-гематолог, при необходимости – генетик, токсиколог, ревматолог, нарколог. Опорными диагностическими критериями выступают:
- Гемограмма. Характерные изменения периферической крови включают снижение Hb и ЦП, возможна ретикулоцитопения. В мазке крови обнаруживается базофильная зернистость эритроцитов, тельца Паппенгеймера. Эритроциты часто уменьшены в размерах (микроцитарная анемия), имеют аномальную форму.
- Биохимические исследования. Сывороточное железо, уровни ферритина и трансферрина повышены, ОЖСС – снижена. Отмечается непрямая гипербилирубинемия, повышение ЛДГ. Может выявляться дефицит витамина В6. При токсической СБА показано исследование содержания свинца в крови.
- Генодиагностика. Осуществляется при подозрении на наследственную форму сидеробластной анемии. Диагноз считается достоверным при выявлении мутаций в причинных генах.
- Исследование мочи. Исследование порфиринов в суточной моче показывает их повышенную экскрецию. Результаты десфералового теста демонстрируют выведение с мочой значительного количество железа.
- Миелограмма. Цитологическая картина пунктата костного мозга указывает на гиперплазию эритроидного ростка миелопоэза. Более 40% предшественников эритроцитов представлены кольцевидными сидеробластами.
- Биопсия внутренних органов. При биопсии печени в гепатоцитах обнаруживаются депозиты железа, иногда – признаки цирроза. Возможно проведение биопсии селезенки, поджелудочной железы.
Лечение сидеробластной анемии
При токсических и лекарственных формах СБА в первую очередь устраняют провоцирующий фактор (отменяют ЛС, проводят дезинтоксикационную терапию и т.д.). Лекарственная терапия сидеробластной анемии включает назначение следующих препаратов:
- Витамин В6. Пиридоксинзависимые формы (некоторые наследственные, алиментарные, алкогольные) поддаются терапии пиридоксина гидрохлоридом или пиридоксальфосфатом. При положительном отклике проводят длительные курсы большими дозами вит. В6 с последующей поддерживающей витаминотерапией.
- Гормонотерапия. Показана пациентам с пиридоксин-резистентной формой сидеробластной анемии. Назначаются анаболические гормоны, андрогены, эритропоэтин.
- Хелаторная терапия. Введение дефероксамина направлено на удаление избытка железа из организма, уменьшение степени выраженности гемосидероза. Для коррекции перегрузки железом также может быть использована флеботомия.
- Вспомогательная терапия. Включает назначение фолиевой кислоты, антиоксидантов (вит. Е, липоевой кислоты), гепатопротекторов.
Прогноз и профилактика
Прогноз при сидеробластной анемии обусловлен ее этиологией. Лучше поддаются коррекции пиридоксин-зависимые и некоторые приобретенные формы (лекарственные, алиментарные). Существенно снижена продолжительность жизни у лиц с СБА, трансфузионно-зависимыми, рефрактерными к лечению, ассоциированными с лейкемией, синдромом Пирсона. Профилактика, прежде всего, касается вторичных сидеробластных анемий: следует полноценно питаться, принимать витаминные комплексы, не допускать интоксикаций (этанолом, свинцом) и контакта с вредными веществами. Для исключения наследственных СБА рекомендуется генетическое консультирование.
3. Клинический случай X-сцепленной сидеробластной анемии с новой миссенс-мутацией cd518 ttg-ttt (leu518phe) в гене alas2. и соавт.// Гематология и трансфузиология. – 2020.
4. Морфология анемий и гемобластозов/ Макаров И.Ю., Меньщикова Н.В., Дубяга Е.В., Левченко Н.Р. – 2018.
Гемолитическая анемия
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
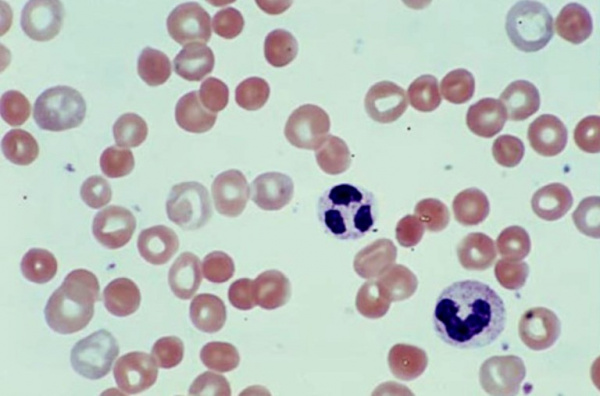
Гемолитическая анемия (ГА) - малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
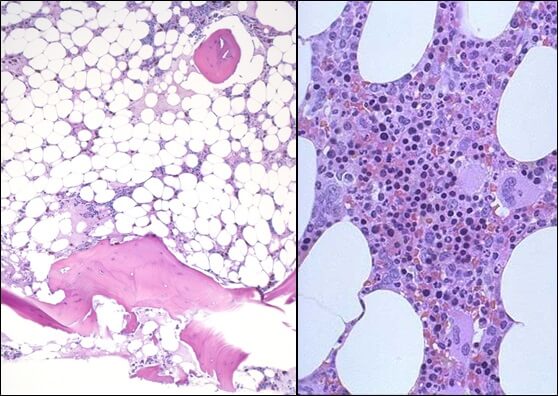
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Центральным звеном патогенеза ГА является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. При аутоиммунном механизме анемии происходит образование антиэритроцитарных АТ (тепловых, холодовых), которые вызывают ферментативный лизис мембраны эритроцитов. Токсические вещества, являясь сильнейшими окислителями, разрушают эритроцит за счет развития метаболических, функциональных и морфологических изменений оболочки и стромы красных кровяных телец. Механические факторы оказывают прямое воздействие на клеточную мембрану. Под влиянием этих механизмов из эритроцитов выходят ионы калия и фосфора, а внутрь поступают ионы натрия. Клетка разбухает, при критическом увеличении ее объема наступает гемолиз. Распад эритроцитов сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии- анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) – обусловлены воздействием антител
- токсические – анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические - анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Симптомы
Наследственные мембранопатии, ферментопении и гемоглобинопатии
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом - обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов – еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические анемии связаны с недостатком определенных ферментов эритроцитов (чаще - Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет. Особенностям клинического течения гемоглобинопатий - талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Приобретенные гемолитические анемии
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия. Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ – при микросфероцитозе, печеночная недостаточность – при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Лечение
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов.
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
4. Клинические рекомендации по диагностике и лечению аутоиммунных гемолитический анемий/ Цветаева Н.В., Никулина О.Ф. - 2014.
Апластическая анемия ( Гипопластическая анемия )
Апластическая анемия – угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
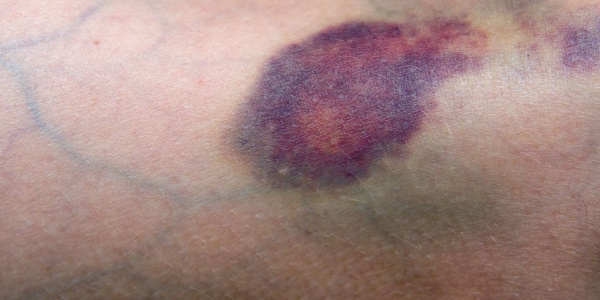
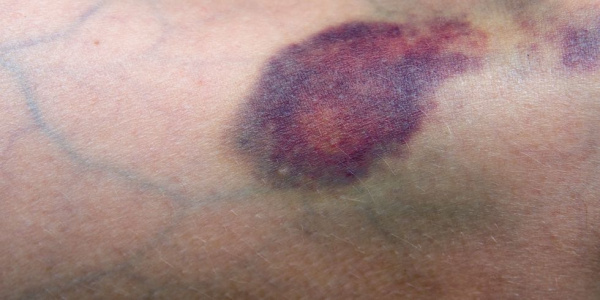
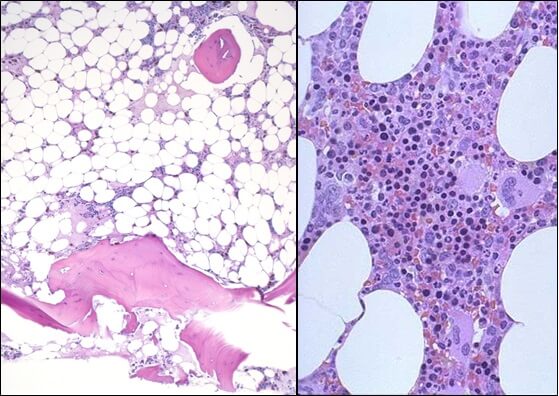
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений – кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.
Читайте также: