Синдром Альпорта: нефрит с нейросенсорной глухотой
Добавил пользователь Владимир З. Обновлено: 08.01.2026
Наследственный нефрит или синдром Альпорта - генетически детерминированная наследственная неиммунная гломерулопатия, проявляющая гематурией (иногда с протеинурией), прогрессирующим снижением почечных функций с развитием хронической почечной недостаточности, часто сочетающаяся с нейросенсорной глухотой и расстройствами зрения.
Прикрепленные файлы: 1 файл
sindrom_alporta.doc
Наследственный нефрит или синдром Альпорта - генетически детерминированная наследственная неиммунная гломерулопатия, проявляющая гематурией (иногда с протеинурией), прогрессирующим снижением почечных функций с развитием хронической почечной недостаточности, часто сочетающаяся с нейросенсорной глухотой и расстройствами зрения.
Впервые заболевание описано в 1902 г. L.G.Guthrie, который наблюдал семью, в нескольких поколениях которой наблюдалась гематурия. В 1915 у членов этой же семьи A.F.Hurst было описано развитие уремии. В 1927 г. A Alport впервые выявил тугоухость у нескольких родственников с гематурией В 50-е годы прошлого века были описаны поражения глаз при подобном заболевании. В 1972 г. у больных с наследственной гематурией при морфологически исследовании почечной ткани Hinglais et al. выявили неравномерное расширение и расслоение гломерулярных базальных мембран. В 1985 г. была идентифицирована генетическая основа наследственного нефрита - мутация в гене коллагена IV типа (Fiengold et al., 1985).
Исследование генетической природы заболевания позволило заключить, что различия в фенотипических проявлениях наследственного нефрита (с тугоухостью или без нее) обусловлены степенью экспрессии мутантного гена. Таким образом, в настоящее время все клинические варианты рассматриваются как проявления одного заболевания и термин "наследственный нефрит" является синонимом термина "синдром Альпорта".
Согласно данным эпидемиологических исследований наследственный нефрит встречается с частотой 17 на 100 000 детского населения.
Генетическая основа болезни - мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики наследственного нефрита.
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена. У большинства больных наследственным нефритом в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения; имеют значение родственные браки между людьми, имеющими одного или более предков, так как в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У детей чаще различают три варианта наследственного нефрита: синдром Альпорта, наследственный нефрит без тугоухости и семейная доброкачественная гематурия.
Синдром Альпорта - наследственный нефрит с поражением слуха. В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома Альпорта расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
Генетической основой развития наследственного нефрита являются мутации в генах альфа-цепей коллагена IV типа. Известно шесть а-цепей IV коллагена Г типа: гены а5- и а6-цепей (Соl4A5 и Соl4A5) находятся на длинном плече Х-хромосомы в зоне 21-22q; гены а3- и а4-цепей (Соl4A3 и Соl4A4) - на 2-й хромосоме; гены a1- и a2-цепей (Соl4A1 и Соl4A2) - на 13-й хромосоме.
В большинстве случаев (80-85%) выявляется Х-сцепленный тип наследования заболевания, связанный с повреждениями гена Соl4A5 в результате делеции, точечных мутаций или нарушений сплайсинга. В настоящее время оги но более 200 мутаций гена Соl4A5, ответственных за нарушение синтеза а5-цепей коллагена IV типа. При этом типе наследования заболевание проявляется у детей обоего пола, но у мальчиков протекает тяжелее.
Мутации в локусах генов Соl4A3 и Соl4A4, ответственных за синтез а3- и а4 - цепей коллагена IV типа, наследуются аутосомно. По данным исследований аутосомно-доминантныи тип наследования отмечается в 16% случаев наследственного нефрита, аутосомно-рецессивный - у 6% больных. Изестно около 10 вариантов мутаций генов Соl4A3 и Соl4A4.
Результатом мутаций является нарушение процессов сборки коллагена IV типа, приводящее к нарушению его структуры. Коллаген IV типа является одним из основных компонентов гломерулярной базальной мембраны, кохлеарного аппарата и хрусталика глаза, патология которых будет выявляться в клинике наследственного нефрита.
Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV) и одной а2-цепи (IV), а также содержит а3, а4, а5-цепи. Наиболее часто при Х-сцепленном наследовании мутация гена Соl4A5 сопровождается отсутствием а3-, а4-, а5- и а6 цепей в структуре коллагена IV типа, а количество о1- и а2-цепей в гломерулярной базальной мембране возрастает. Механизм этого феномена неясен, предполагается, что причиной являются посттранскрипционные изменения мРНК.
Отсутствие а3-, а4- и а5-цепей в структуре IV типа коллагена базальных мембран клубочков приводит к их истончению и ломкости на ранних стадиях синдрома Альпорта, что клинически проявляется чаще гематурией (реже гематурией с протеинурией или только протеинурией), снижением слуха и лентиконусом. Дальнейшее прогрессирование заболевания приводит к утолщению и нарушению проницаемости базальных мембран на поздних стадиях заболевания, с разрастанием в них коллагена V и VI типов, проявляющихся в нарастании протеинурии и снижении почечных функций.
Характер мутации, лежащей в основе наследственного нефрита, во многом определяет его фенотипическое проявление. При делеции Х-хромосомы с одновременной мутацией генов Соl4A5 и Соl4A6, ответственных за синтез а5- и а6-цепей коллагена IV типа, синдром Альпорта сочетается с лейомиоматозом пищевода и половых органов. По данным исследований при мутации гена Соl4A5, связанной с делецией, отмечаются большая тяжесть патологического процесса, сочетание почечного поражения с экстраренальными проявлениями и ранним развитием хронической почечной недостаточности, по сравнению сточечной мутацией этого гена.
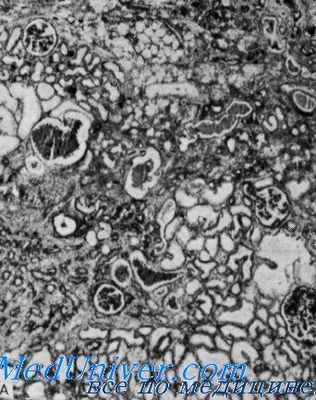
Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие электронно-плотных гранул. Поражение гломерул может быть неоднородным у одного и того же больного, от минимального фокального поражения мезангия до гломерулосклероза. Гломерулит при синдроме Альпорта всегда носит иммунонегативный характер, что отличает его от гломерулонефрита. Характерны развитие атрофии канальцев, лимфогистиоцитарная инфильтрация, наличие "пенистых клеток" с включениями липидов - липофагами. При прогрессировании заболевания выявляется утолщение и выраженная деструкция базальных мембран клубочков.
Выявляются определенные сдвиги в состоянии имунной системы. У больных наследственным нефритом отмечено снижение уровня Ig A и склонность к повышению концентрации IgМ в крови, уровень IgG может быть повышен на ранних стадиях развития заболевания и снижаться на поздних сроках. Возможно, повышение концентрации IgM и G является своеобразной компенсаторной реакцией в ответ на дефицит IgA.
Функциональная активность системы Т-лимфоцитов снижена; отмечается избирательное снижение В-лимфоцитов, ответственных за синтез Ig A, нарушается фагоцитарное звено иммунитета, в основном за счет нарушения процессов хемотаксиса и внутриклеточного переваривания в нейтрофилах
При исследовании биоптата почек у больных с синдромом Альпорта по данным электронной микроскопии наблюдаются ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях наследственного нефрита дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при наследственном нефрите - расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
Выделяют три варианта наследственного нефрита
I вариант - клинически проявляется нефритом с гематурией, тугоухостью ипоражением глаз. Течение нефрита прогрессирующее с развитием ХПН. Тип наследования - доминантный, сцепленный с Х-хромосомой. Морфологически выявляется нарушение структуры базальной мембраны, ее истончение и расщепление.
II вариант- клинически проявляется нефритом с гематурией без тугоухости. Течение нефрита прогрессирующее с развитием хронической почечной недостаточности. Тип наследования - доминантный, сцепленный с Х-хромосомой. Морфологически выявляется истончение базальной мембраны капилляров клубочков (особенно laminadensa).
III вариант - доброкачественная семейная гематурия. Течение благоприятное, хроническая почечная недостаточность не развивается. Тип наследования - аутосомно-доминантный или аутосомно-рецессивный. При аутосомно-рецессивном типе наследования у женщин отмечено более тяжелое течение заболевания.
Первые симптомы синдрома Альпорта в виде изолированного мочевого синдрома чаще выявляются у детей первых трех лет жизни. В большинстве случаев заболевание обнаруживается случайно. Мочевой синдром выявляется при профилактическом обследовании ребенка, перед поступлением в детское учреждение или во время ОРВИ. В случае появления патологии в моче во время ОРВИ. При наследственном нефрите в отличие от приобретенного гломерулонефрита отсутствует латентный период.
В начальной стадии болезни самочувствие ребенка страдает мало, характерной особенностью является упорство и стойкость мочевого синдрома. Одним из основных признаков является гематурия различной степени выраженности, наблюдаемая в 100% случаев. Усиление степени гематурии отмечается во время или после инфекций дыхательных путей, физической нагрузки или после профилактических прививок. Протеинурия в большинстве случаев не превышает 1 г/сут, в начале заболевания может быть непостоянной, по мере прогрессирования процесса протеинурия нарастает. Периодически в мочевом осадке может присутствовать лейкоцитурия с преобладанием лимфоцитов, что связывают с развитием интерстициальных изменений.
В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного: появляются интоксикация, мышечная слабость, артериальная гипотония, часто нарушение слуха (особенно у мальчиков), иногда нарушение зрения. Интоксикация проявляется бледностью, утомляемостью, головными болями. В начальной стадии болезни снижение слуха в большинстве случаев выявляется только с помощью аудиографии. Снижение слуха при синдроме Альпорта может возникнуть в различные периоды детства, однако чаще всего тугоухость диагностируется в возрасте 6-10 лет. Начинается снижение слуха у детей с высоких частот, достигая значительной степени при воздушном и костном проведении, переходя из звукопроводящей в звуковоспринимающую тугоухость. Снижение слуха может быть одним из первых симптомов заболевания и может предшествовать мочевому синдрому.
В 20% случаев у больных с синдромом Альпорта отмечаются изменения со стороны органов зрения. Наиболее часто выявляются аномалии со стороны хрусталика: сферофокия, лентиконус передний, задний или смешанный, разнообразные катаракты. В семьях с синдромом Альпорта наблюдается значительная частота миопии. Ряд исследователей постоянно в этих семьях отмечают билатеральные перимакулярные изменения в виде ярких беловатых или желтоватых грануляций в области желтого тела. Они считают этот признак постоянным симптомом, который имеет высокую диагностическую ценность при синдроме Альпорта. К. S. Chugh и соавт. (1993) при офтальмологическом исследовании выявили у больных синдромом Альпорта снижение остроты зрения в 66,7% случаев, передний лентиконус - в 37,8%, пятна на сетчатке - в 22,2%, катаракту - в 20%, кератоконус - в 6,7%.
У части детей при наследственном нефрите, особенно при формировании почечной недостаточности, отмечают существенное отставание в физическом развитии. По мере прогрессирования почечной недостаточности развивается артериальная гипертензия. У детей она чаще выявляется в подростковом периоде и в более старших возрастных группах.
Характерно наличие у больных с наследственным нефритом разнообразных (более 5-7) стигм соединительнотканного дизэмбриогенеза. Среди соединительнотканных стигм у больных наиболее часто встречаются гипертелоризм глаз, высокое нёбо, аномалии прикуса, аномальная форма ушных раковин, искривление мизинца на руках, «сандалевидная щель» на стопах. Для наследственного нефрита характерна однотипность стигм дизэмбриогенеза внутри семьи, а также высокая частота их распространения у родственников пробандов, по линии которых передается болезнь.
На ранних этапах болезни выявляется изолированное снижение парциальных функций почек: транспорта аминокислот, электролитов, концентрационной функции, ацидогенеза, в дальнейшем изменения касаются функционального состояния как проксимального, так и дистального отдела нефрона и носят характер сочетанных парциальных нарушений. Снижение клубочковой фильтрации наступает позднее, чаще в подростковом периоде. По мере прогрессирования наследственного нефрита развивается анемизация.
Таким образом, для наследственного нефрита характерна стадийность течения болезни: сначала латентная стадия или скрытых клинических симптомов, проявляющаяся минимальными изменениями мочевого синдрома, затем наступает постепенная декомпенсация процесса со снижением почечных функций с манифестными клиническими симптомами (интоксикация, астенизация, отставание в развитии, анемизация). Клинические симптомы появляются обычно вне зависимости от наслоения воспалительной реакции.
Синдром Альпорта: нефрит с нейросенсорной глухотой
Синдром Альпорта: нефрит с нейросенсорной глухотой
В 1927 г. Alport описал встречающийся преимущественно у мужчин синдром хронического нефрита с интермиттирующей или массивной гематурией, прогрессирующей. почечной недостаточностью и прогрессирующей нейросеисорной глухотой. С тех пор вышло несколько сот публикаций, посвященных синдрому Альпорта, составляющему, как считают, около 1% среди наследственной глухоты.
Клинические данные. Почки. Частым синдромом является гематурия. В течение нескольких лет она бывает выражена резко, а затем ослабевает. Гематурия может выявиться на первом году жизни («красная пеленка») в сочетании с асимптоматической альбуминурией и пиурией, но чаще она появляется в течение первого или второго десятилетия жизни. Гипертензия и почечная недостаточность встречаются главным образом у мальчиков в возрасте от 13 до 19 лет.
Больные мужчины обычно умирают до 30-летнего возраста вследствие медленно прогрессирующей уремии. У женщин заболевание почек выражено менее тяжело, но отмечается тенденция к возникновению тяжелой токсемии при беременности (Crawfurd, Toghill, Chiricosta at al.). He являются редкостью и спонтанные аборты (около 15%). Однако продолжительность жизни у больных женщин обычно нормальная.
Орган зрения. Глазная патология отмечается примерно у 10% больных с синдромом Альпорта. Наблюдаются коническая форма хрусталика, сферофакия и особенно часто кортикальные катаракты (Sohar, Perrian, Arnott, Crawfurd, Toghill, Arenberg et al., Purriel et al., Hauser).
Орган слуха. Степень глухоты варьирует. Во втором десятилетии жизни обычно выявляется симметричная прогрессирующая нейросенсорная глухота в основном на средние и высокие частоты. Дефект слуха относительно легкий и помещать больных в специальные школы для глухих приходится редко (Klotz, Johnson, Hagan, Gekle et al.).
Дефект слуха чаще наблюдается у мальчиков. Он встречается также и у девочек, но в меньшей пропорции. Cassidy с соавт., изучив 7 семей с синдромом Альпорта, обнаружил глухоту примерно у 55% мужчин и у 40% женщин. Аналогичные данные сообщили Chiricosta с сотр.. Слабость восприятия низких тонов была отмечена Ferguson и Rance и Hauser.
Spear с сотр. произвели аудиометрическое исследование 16 больным из четырех семей. У детей младше 10-летнего возраста слух обычно был нормальным. Начиная со второго десятилетия жизни, выявлялась нейросенсорная глухота около 50 дБ, выраженная резче на высоких частотах. Различение речи обычно было нормальным. SISI-тест в 3 из 7 случаев был положительным, в то же время tone-decay-тест был положительным только в 1 случае из 9. Такие изменения, обнаруженные также Miller с соавт., позволяют предположить кохлеарное происхождение глухоты.
Вестибулярная система. Калорическая стимуляция вызнала сниженную реакцию (Miller et al., Celes-Blaubach et al.).
Лабораторные данные. У больных с тяжелым течением почечного заболевания при помощи внутривенной пиелографии была обнаружена атрофия некоторых участков почек.
Анализ мочи показал различную степень гематурии, протеинурии и пиурии. В моче могут быть обнаружены клеточные слепки, содержащие красные кровяные шарики. Параллельно тяжести заболевания повышается уровень мочевого азота в крови.
Синдром Альпорта: нефрит с нейросенсорной глухотой
Синдром Альпорта
Наследственный нефрит (синдром Альпорта) - генетически детерминированная наследственная неиммунная гломерулопатия, проявляющая гематурией (иногда с протеинурией), прогрессирующим снижением почечных функций с развитием хронической почечной недостаточности , часто сочетающаяся с нейросенсорной глухотой и расстройствами зрения.
Причиной синдрома Альпорта является дефектный ген в Х-хромосоме. У женщин с дефектным геном в одной из их двух Х-хромосом обычно нет симптомов, хотя их почки могут работать менее эффективно, чем нормальные. У мужчин с дефектным геном (у мужчин нет второй Х-хромосомы, чтобы компенсировать этот дефект) обычно развивается почечная недостаточность в возрасте между 20 и 30 годами.
КЛИНИКА
Отмечается выраженный клинический полиморфизм. Наиболее характерным является гематурия, протеинурия, периодическая бактериурия, снижение слуха. Первые признаки синдрома Альпорта появляются обычно в возрасте 5-10 лет. Выраженность мочевого синдрома поначалу минимальна, нет нарушений функции почек. В дальнейшем постепенно появляются и неуклонно нарастают явления ХПН (более быстро и тяжело у лиц мужского пола). Снижение слуха может развиться еще до появления почечной патологии. В начале это нейросенсорное снижение слуха высоких тонов, далее -- низких, переходящее из звукопроводящей в звуковоспринимающую тугоухость. Могут быть также миастения, потеря памяти и интеллекта, тромбоцитопения. В семьях больных описаны лица с изолированной тугоухостью, доброкачественной гематурией.
ДИАГНОЗ
Важно составление родословной и обнаружение лиц как с сочетанием нефрита и глухоты, так и с доброкачественной гематурией, тухоухостью, ХПН. Диагноз подтверждают при биопсии почек, обнаружении в моче D/L-3-гидроксипролина, глюкозилгалактозилоксилизина. У части больных выявляют сужение прилоханочного отдела мочеточника, удвоение, незавершенный поворот почек.
ЛЕЧЕНИЕ
Назначение глюкокортикоидов и цитостатиков неэффективно и лишь ухудшает прогноз. Диета высококалорийная в соответствии с возрастом и функциональным состоянием почек. Показано раннее выявление и активное лечение как мочевой инфекции, так и хронических очагов инфекции, последовательные курсы "трофических" препаратов -- АТФ, кокарбоксилазы, пиридоксина, ксидифона, витаминов А, Е, В5, В15 etc., фитотерапия. Некоторые авторы описывают положительный эффект от применения делагила в дозе 5-10 мг/кг в сутки в течение 6-12 мес, левамизола (2,5 мг/кг в сутки 2-3 раза в неделю в течение 4-6 нед). При начальных признаках ХПН показана трансплантация почки, хотя у 1-5% после трансплантации может развиться нефрит с антителами к базальной мембране клубочков.
Наиболее эффективным методом лечения наследственного нефрита является своевременная трансплантация почки.
Синдром Альпорта
Синдром Альпорта - генетически гетерогенное заболевание, характеризующееся нефритическим синдромом (т.е. гематурией, протеинурией, гипертонией, в конечном итоге почечной недостаточностью), часто с нейросенсорной глухотой и, реже, офтальмологическими симптомами. В основе заболевания лежит генная мутация, повреждающая коллаген IV типа. Диагностика производится на основании данных анамнеза, включая семейный анамнез, результатов анализа мочи и биопсии (почки или кожи). Лечение такое же, как и при хронической болезни почек, иногда включает пересадку почки Трансплантация почки Трансплантация почек является наиболее распространенным видом трансплантации цельных органов. (См. также Обзор трансплантации (Overview of Transplantation)). Основное показание для трансплантации. Прочитайте дополнительные сведения .
Синдром Альпорта – это нефритический синдром, вызванный мутацией гена COL4A3, COL4A4, COL4A5, (кодирующего альфа-5 цепь коллагена IV типа), что приводит к формированию измененных коллагеновых волокон IV типа. Механизм развития гломерулярной болезни в результате изменений структуры коллагена неизвестен, но предполагают, что в основе лежит нарушение структуры и функции; у членов большинства семей развивается утолщение и истончение гломерулярных и канальцевых базальных мембран с образованием многослойной плотной пластинки с очаговым или местным распределением (пример переплетения "плетенка"). В результате происходит рубцевание гломерулярной ткани и интерстициальный фиброз.
Болезнь обычно наследуется по Х-сцепленному механизму, хотя существуют аутосомно-рецессивные и, редко, аутосомно-доминантные варианты. Случаи наследования по Х-хромосоме могут быть клинически классифицированы как:
Ювенильная форма: почечная недостаточность развивается между 20 и 30 годами
Зрелая (взрослая) форма: почечная недостаточность развивается у людей > 30 лет
Клинические проявления
Классическое Х-хромосомное заболевание у мужчин и аутосомно-рецессивное заболевание клинически сходны. У пациентов развиваются почечные симптомы, характерные для острого нефритического синдрома (например, микрогематурия, гипертензия, а впоследствии макрогематурия с протеинурией), а прогрессирование заболевания до почечной недостаточности происходит в возрасте 20–30 лет (ювенильные формы).
Нейросенсорная тугоухость встречается часто, при этом характерно нарушение восприятия более высоких частот; она может оставаться недиагностированной в раннем детстве.
Реже, чем тугоухость, наблюдаются офтальмологические патологии: катаракта (чаще всего), передний лентиконус (простая протрузия конуса к переднему полюсу хрусталика из-за утолщения капсулы хрусталика), сферофакия (сферическая деформация хрусталика, предрасполагающая к подвывиху хрусталика), нистагм, пигментный ретинит, слепота.
Х-хромосомное заболевание встречается у гетерозиготных женщин, которые, потому что они имеют одну нормальную Х-хромосому, как правило, имеет менее тяжелые, более медленно прогрессирующие симптомы, чем у мужчин.
У некоторых мужчин с Х-хромосомным заболеванием почечная недостаточность развиваются после 30 лет с потерей слуха, которая происходит с опозданием или в легкой форме, а аутосомно-доминантное заболевание, как правило, не приводит к почечной недостаточности до возраста ≥ 45 лет (взрослые формы).
У больных с Х-хромосомным синдромом Альпорта, сенсоневральная потеря слуха, обычно, проявляется в детсве, в то время, как болезнь почек часто не проявляется до взрослого возраста.
Синдром Альпорта ( Гематурический нефрит , Наследственный нефрит 1 типа , Семейный гломерулонефрит )
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
МКБ-10
Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН. У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам. Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
1. Наследственный нефрит (синдром Альпорта) / Сунгатуллина И.Л. // Казанский медицинский журнал – 2002 – Т.83, №1.
2. Проект клинических рекомендаций по диагностике и лечению синдрома Альпорта у детей / Длин В.В., Игнатова М.С., Конькова Н.Е. – 2014.
3. Наследственные заболевания почек, протекающие с гематурией / Длин В.В., Игнатова М.С. // Российский вестник перинатологии и педиатрии – 2014 - №3.
Читайте также: