Синдром IPEX
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
Modern Pediatrics. Ukraine. (2022). 2(122): 63-71. doi 10.15574/SP.2022.122.63
Шадрин О. Г. 1 , Марушко Т. Л. 1 , Волоха А. П. 2 , Марушко Р. В. 1
1 ГУ «Институт педиатрии, акушерства и гинекологии имени академика Е.М. Лукьяновой НАМН Украины», г. Киев
2 Национальный университет охраны здоровья Украины имени П.Л. Шупика, г. Киев
Для цитирования: Shadrin OG, Marushko TL, Volokha AP, Marushko RV. (2022). Primary immunodeficiency: IPEX-syndrome. Literature review and clinical case. Modern Pediatrics. Ukraine. 2(122): 63-71. doi 10.15574/SP.2022.122.63.
Статья поступила в редакцию 29.11.2021 г., принята в печать 06.03.2022 г.
IPEX-синдром (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome) — тяжелое наследственное Х-сцепленное заболевание группы первичных иммунодефицитов, ассоциированное с мутациями в гене FOXP3, кодирующем ключевой фактор транскрипции для Т-регуляторных лимфоцитов. В патогенезе заболевания основную роль играет нарушение дозревания CD25+CD4+-Т-регуляторных лимфоцитов (TR), осуществляющих отрицательную селекцию аутореактивных клонов Т- и В-лимфоцитов, вследствие чего происходит потеря организмом аутотолерантности и раннее развитие полиорганной аутоиммунной патологии в сочетании с выраженным нарушением противовирусного и антибактериального иммунитета. Типичной клинической картиной IPEX-синдрома является триада: аутоиммунная энтеропатия, эндокринопатия (сахарный диабет 1-го типа, аутоиммунный тиреоидит), поражение кожи и/или слизистых оболочек, однако возможной является любая комбинация аутоиммунной патологии или изолированное поражение отдельных органов. При этом поражение кишечника (аутоиммунная энтеропатия) выявляется практически у 100% пациентов. Лабораторные иммунологические маркеры IPEX-синдрома отсутствуют (заболевание может быть выявленным только при помощи генетического обследования — панельного секвенирования генов), что наряду с вариабельностью клинических проявлений создает значительные диагностические трудности.
Представлен клинический случай IPEX-синдрома, клиническими особенностями которого были изолированная энтеропатия без клинико-лабораторных признаков аутоиммунного поражения других органов, а также тяжелая степень поражения слизистой оболочки тонкой кишки (тотальная атрофия ворсин) и стремительное прогрессирование заболевания, несмотря на поздний дебют клинических проявлений (на 2-м году жизни ребенка), ассоциируемый с «мягкими» мутациями гена, более легким течением и более благоприятным прогнозом. Лабораторными особенностями данного случая были: отрицательные серологические маркеры аутоиммунного поражения кишечника и повышенный уровень Т-регуляторных лимфоцитов CD4+ CD25+ CD127 low+, хотя для IPEX-синдрома характерны снижение или отсутствие Т-регуляторных клеток.
IPEX-синдром у данного пациента подтвержден генетическим исследованием — при секветировании гена FOXP3 выявлена патогенная мутация с.736-2А>Т (Splice acceptor).
Исследование выполнено в соответствии с принципами Хельсинкской декларации. На проведение исследований получено информированное согласие родителей.
Авторы заявляют об отсутствии конфликта интересов.
Ключевые слова: дети раннего возраста, первичный иммунодефицит, IPEX-синдром, мутации гена FOXP3, аутоиммунная энтеропатия, диагностика.
ЛІТЕРАТУРА
8. Чубарова АИ, Шумилов ПВ, Костомарова ЕА, Хаматвалеева ГР, Дмитриева ЮА. (2016). Клинический случай синдрома иммунной дисрегуляции, полиэндокринопатии (IPEX-синдрома) с изолированным поражением кишечника. Педиатрия. 95 (6): 187-192.
--> --> --> -->Синдром IPEX
Эндокринологический научный центр Минздравсоцразвития России
Морозовская детская городская клиническая больница
Морозовская детская городская клиническая больница
Морозовская ДГКБ Департамента здравоохранения Москвы
Эндокринологический научный центр, Москва
Х-сцепленные иммунная дисрегуляция, полиэндокринопатия и энтеропатия (IPEX-синдром): описание клинического случая и краткий обзор литературы
Журнал: Проблемы эндокринологии. 2014;60(5): 29‑33
Эндокринологический научный центр Минздравсоцразвития России
IPEX-синдром (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) является одной из редких наследственных Х-сцепленных форм неонатального сахарного диабета, ассоциированного с мутациями в гене FOXP3. Заболевание характеризуется сочетанием полиэндокринопатии (чаще неонатальный сахарный диабет) с энтеропатией и иммунной дисрегуляцией. Прогноз в большинстве случаев неблагоприятный. Представлено первое в отечественной литературе описание генетически подтвержденного клинического случая IPEX-синдрома.
Эндокринологический научный центр Минздравсоцразвития России
Морозовская детская городская клиническая больница
Морозовская детская городская клиническая больница
Морозовская ДГКБ Департамента здравоохранения Москвы
Эндокринологический научный центр, Москва
IPEX-синдром (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) - тяжелое наследственное Х-сцепленное заболевание, ассоциированное с мутациями в гене FOXP3 (forkhead box protein 3 gene), кодирующем ключевой фактор транскрипции для Т-регуляторных лимфоцитов. Дисфункция Т-регуляторных лимфоцитов вызывает развитие полиорганной аутоиммунной патологии в сочетании с выраженным дефектом противовирусного и антибактериального иммунитета, что в большинстве случаев приводит к летальному исходу в течение 1 года жизни от генерализованного сепсиса или тяжелой мальабсорбции, осложненной кишечными кровотечениями. Фатальные исходы нередко ассоциированы с вакцинацией, вирусными инфекциями и другими экзогенными иммуностимулирующими воздействиями. В то же время в литературе описаны взрослые пациенты с неполной формой IPEX-синдрома с мягким течением [1].
Частота встречаемости IPEX-синдрома в настоящее время не определена. По данным О. Rubio-Cabezas [1], мутации в гене FOXP3 выявлены у 4% пациентов мужского пола с перманентным неонатальным сахарным диабетом (ПНСД). Женщины являются носителями мутации и, как правило, здоровы.
На сегодняшний день насчитываются более 136 пациентов с генетически подтвержденным IPEX-синдромом [2]. В отечественной литературе случаи сахарного диабета (СД), связанные с мутациями в гене FOXP3, ранее не были описаны.
Клинический случай
Мальчик от второй беременности, протекавшей на фоне раннего токсикоза. Роды на 37-й неделе, масса тела при рождении 3360 г, длина тела 53 см. Оценка по шкале Апгар 7-8 баллов. Родители и старший ребенок в семье (девочка 5 лет) здоровы.
С рождения отмечены явления гиперкератоза, пластинчатого шелушения кожи, иктеричность кожи и склер, неврологическая симптоматика в виде синдрома возбуждения ЦНС. На 5-е сутки жизни переведен в отделение патологии новорожденных с подозрением на гемолитическую болезнь новорожденных по системе АВ0 [у матери I (0) группа крови Rh+; у ребенка III (В) группа крови Rh+]. В анализах крови: анемия (Hb 95 г/л), эозинофилия (11-20%), гипербилирубинемия (общий билирубин 209,8 мкмоль/л, непрямой 199 мкмоль/л), гипопротеинемия и гипоальбуминемия. Выписан по настоянию родителей на 20-е сутки жизни на фоне частичного улучшения состояния и сохраняющейся эозинофилии.
В возрасте 29 дней повторная госпитализация в связи с нарастанием вялости, потерей массы тела, появлением субфебрилитета. При поступлении выявлено повышение гликемии до 28 ммоль/л, метаболический ацидоз (рН крови 6,9, ВЕ –25,7 ммоль/л). Диагностирован сахарный диабет и начата интенсивная терапия, включающая инсулинотерапию.
Несмотря на нормализацию КЩС и достижение субкомпенсации гликемии, состояние ребенка оставалось тяжелым. Отмечались вялость, снижение аппетита, признаки инфицированного атопического дерматита, субфебрилитет, эпизодически диарея, отсутствовала прибавка массы тела. В анализах крови: нейтрофильный лейкоцитоз (16-22·10 9 /л), эозинофилия (10%), анемия (гемоглобин 76 г/л, эритроциты 2,5·10 12 /л), гипопротеинемия (53 г/л), гипоальбуминемия (33 г/л); повышение уровня печеночных трансаминаз. Уровень прокальцитонина как маркера тяжелой бактериальной или грибковой инфекции составил 4 нг/мл (норма 0,05-0,1 нг/мл); при этом очаг воспаления установить не удалось. HbA1c был умеренно повышен (6,4%), при этом имело место снижение уровня С-пептида до 21 пмоль/л (норма 80-850) и отсутствовали антитела к инсулину, островковым клеткам, тирозинфосфатазе и глутаматдегидрогеназе. В тиреоидном профиле: ТТГ 8,85 мМЕ/л, св. Т4 12,8 пмоль/л, АТ ТПО 111 ед/л (норма 0-34).
На 40-е сутки пребывания в стационаре, несмотря на проводимую терапию, включая антибиотики резерва, состояние ребенка резко ухудшилось, появились фебрильная лихорадка, явления токсикоза с эксикозом, метаболический ацидоз (рН 7,0 BE –27,0), при этом сохранялась субкомпенсация углеводного обмена (гликемия до 12,6 ммоль/л). Интенсивная терапия оказалась безрезультатной, и на фоне нарастающих явлений полиорганной недостаточности в возрасте 10 нед был зафиксирован летальный исход.
Учитывая наличие у мальчика неонатального СД в сочетании с тяжелой резистентной к терапии инфекцией, энтеритом, поражением кожи, аутоиммунным тиреоидитом, эозинофилией, был заподозрен IPEX-синдром.
Молекулярно-генетические исследования
Геномную ДНК выделяли из периферических лейкоцитов с использованием стандартных методов. С помощью полимеразной цепной реакции (ПЦР) амплифицировали фрагменты геномной ДНК, охватывающие кодирующую последовательность гена FOXP3 с примыкающими участками интронов. После электрофореза в 1% агарозном геле продукты ПЦР очищали с использованием набора Cleanup Standard («Евроген», Россия), а затем секвенировали на автоматическом секвенаторе ABI Genetic Analyzer 3130 («Applied Biosystems», США). При проведении ПЦР и последующем секвенировании соответствующих экзонов и примыкающих участков интронов использовали следующие олигонуклеотиды:
1F, 5’-CTCAGGTGGTCGAGTATCTC-3’; 3R, 5’-TTTGACCCCCAGAGTACTG-3’; 5R, 5’-GATGAAGCCTGAGCTGAGATC-3’; 6F, 5’-TGGGGCTCAGAGGAGAGAAC-3’; 8R, 5’-GGCAGCATGGAGCTCCTTTG-3’; 9F, 5’-GTGAGATCTCAGGCCTGTAG-3’; 11R, 5’-CAGTGGAAACCTCACTTCTTG-3’.
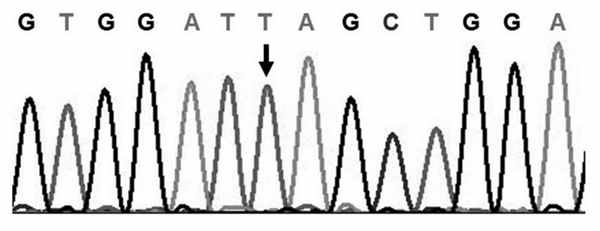
При секвенировании гена FOXP3 у ребенка выявлена гемизиготная трансверсия c.1228G>T в экзоне 11, что приводило к замене кодона глутаминовой кислоты (GAG) на стоп-кодон (TAG) в положении 410 (p.E410X) (см. рисунок, а). Рисунок 1. Результаты генетического исследования. а - фрагмент последовательности экзона 11 гена FOXP3 у пациента. Гемизиготная трансверсия c.1228GмT (отмечена стрелкой) в экзоне 11 с заменой кодона глутаминовой кислоты (GAG) на стоп-кодон (TAG) в положении 410 (p.E410X) У матери ребенка данная мутация была выявлена в гетерозиготном состоянии (см. рисунок, б). Рисунок 2. Результаты генетического исследования. б - гетерозиготная мутация в том же положении (отмечена стрелкой) у матери пациента. Данная мутация описывается впервые.
IPEX-синдром был впервые клинически описан в 1982 г. B. Powell и соавт. [4]. В 2000-2001 гг. было доказано, что в основе заболевания лежит дефект гена FOXP3 7.
FOXP3 локализован на хромосоме Xp11.23, состоит из 11 кодирующих экзонов и специфически экспрессируется СD4+СD25+ регуляторными Т-клетками в тимусе и на периферии [8]. Ген FOXP3 кодирует специфический фактор транскрипции FOXP3 (forkhead box protein 3) или скарфин, отвечающий за образование и функцию регуляторных Т-лимфоцитов СD4+СD25+, обладающих иммуносупрессивными свойствами [9, 10]. Иммуносупрессивные CD4+CD25+ регуляторные Т-клетки составляют небольшой процент (5-10%) от популяции СD4 Т-лимфоцитов (T-хелперов), которые развиваются в тимусе. Данные клетки экспрессируют рецептор α-цепи интерлейкина 2 (CD25), цитотоксический Т-лимфоцит-ассоциированный антиген 4 (CTLA-4) и глюкокортикоид-индуцируемый рецептор TNF (GITR), а также продуцируют иммуносупрессивные цитокины ИЛ-10 и TGF-β [9, 10]. Фактор транскрипции FOXP3 - наиболее специфический молекулярный маркер CD4+CD25+регуляторных Т-клеток [8].
Наличие мутаций в гене FOXP3 обусловливает повышение иммунологической реактивности организма и многократно увеличивает риск развития аутоиммунных заболеваний. Скарфин состоит из 431 аминокислоты и имеет несколько функционально значимых доменов, включающих С-концевой ДНК-связывающий домен (forkhead box); N-концевой домен; домены «цинковых пальцев» и «лейциновой молнии», участвующие в межбелковых взаимодействиях.
В настоящее время в гене FOXP3 описано более 63 мутаций [2]. Большинство из них расположены в экзонах 9-11, кодирующих С-терминальный ДНК-связывающий домен, и в экзонах 1-5, кодирующих N-терминальный домен [2]. Наиболее характерными для пациентов с IPEX-синдромом являются нонсенс-мутации и мутации со сдвигом рамки считывания; реже встречаются миссенс-, сплайсинг-мутации и мутации, приводящие к нарушению полиаденилирования мРНК [2, 11, 12]. Мутация, обнаруженная у нашего пациента, относится к нонсенс-мутациям и локализована в экзоне 11 гена FOXP3. Характер и локализация мутации определяют тяжесть клинических проявлений заболевания. Нонсенс-мутации, а также сплайсинг-мутации, мутации со сдвигом рамки считывания и миссенс-мутации, расположенные в ДНК-связывающем С-терминальном домене, приводят к полной потере функции протеина FOXP3 и ассоциированы с тяжелой (классической) формой IPEX-синдрома [2, 13]. Для пациентов с миссенс-мутациями, расположенными вне ДНК-связывающего сайта, а также с мутациями, вызывающими нарушение полиаденилирования мРНК, характерно сохранение остаточной функции скарфина и более мягкое течение заболевания [2, 14].
В большинстве случаев IPEX-синдром дебютирует сразу после рождения или в течение первого года жизни пациентов [1, 2, 15]. Клинические проявления IPEX-синдрома крайне вариабельны. Для большинства пациентов характерны нормальные массо-ростовые показатели при рождении, но вскоре развивается классическая триада признаков, включающая аутоиммунную энтеропатию (100% случаев), полиэндокринопатию (70-80%) и поражения кожи и слизистых (около 65%) [1]. Тяжелые рецидивирующие инфекции также являются одним из ведущих признаков заболевания и нередко приводят к развитию генерализованного сепсиса и летальному исходу в течение первых 2 лет жизни пациентов [1, 2, 16]. Аутоиммунная энтеропатия - один из наиболее постоянных признаков IPEX-синдрома - манифестирует в первые месяцы жизни ребенка и характеризуется тяжелым прогрессирующим течением с развитием синдрома мальабсорбции, приводящем к тяжелой белково-энергетической недостаточности. Морфологические изменения характеризуются умеренной или выраженной атрофией ворсин с мононуклеарной инфильтрацией собственной пластины слизистой оболочки тонкой кишки.
В ряде случаев тотальная атрофия ворсин ассоциирована с некрозом эпителиальных клеток и формированием криптабсцессов. При иммунологическом исследовании определяются антитела к антигенам энтероцитов: AIE-75 и виллину [17].
Кожный синдром может быть представлен эксфолиативным дерматитом, ихтиозоформным дерматитом или псориазом. У некоторых пациентов описаны тяжелые хейлиты, ониходистрофия, алопеция. Кожные проявления чаще всего диффузны, устойчивы к терапии антигистаминными препаратами и кортикостероидами местного действия; часто осложняются вторичной бактериальной или грибковой инфекцией [18].
Эндокринопатии в большинстве случаев представлены аутоиммунным инсулинзависимым СД, дебютирующим в течение первых 6 мес жизни ребенка. Гораздо реже нарушения углеводного обмена выявляются у детей старшего возраста [15, 19]. Аутоиммунный тиреоидит - вторая по частоте эндокринная патология у пациентов с IPEX-синдромом - чаще всего проявляется гипотиреозом [1]. Как правило, определяется повышение уровня антител к тиреопероксидазе и тиреоглобулину.
Эффективная терапия IPEX-синдрома в настоящее время, к сожалению, не разработана. Лечение пациентов проводится посиндромно; большинство детей нуждаются в парентеральном питании. Некоторые авторы описывают положительный эффект иммуносупрессивных препаратов (циклоспорин А, такролимус, сиролимус и т.д.), которые позволяют частично контролировать аутоиммунные нарушения [24, 25]. Наиболее перспективным направлением в терапии IPEX-синдрома считается пересадка костного мозга [11, 26].
Заключение
Представленный клинический случай демонстрирует не только классическое течение IPEХ-синдрома, но и трудности проведения дифференциальной диагностики заболевания у детей раннего возраста в связи с неспецифичностью клинической симптоматики и редкостью данной патологии. Молекулярно-генетический анализ гена FOXP3 показан всем пациентам мужского пола с инсулинзависимым СД, дебютировавшим в течение 1 года жизни, при наличии сочетанной аутоиммунной патологии.
IPEX-синдром
IPEX-синдром – это редкое тяжелое наследственное заболевание, которое характеризуется нарушением функций иммунной системы, аутоиммунным поражением эндокринных органов и кожи. Клинически наиболее часто проявляется кожными высыпаниями, напоминающими экзему или псориаз, сахарным диабетом, профузной диареей. Практически любые инфекции сразу становятся генерализованными вплоть до развития септического состояния. Окончательный диагноз ставится после проведения генетического исследования. Основной метод лечения – пересадка стволовых клеток. Осуществляется иммуносупрессивная, гормонозаместительная и симптоматическая терапия.
МКБ-10
Общие сведения
Причины
Заболевание возникает вследствие мутации в гене FOXP3, локализующемся в локусе Xp11.23. Это ген кодирует образование белка скарфина. Скарфин отвечает за созревание и функционирование особой группы Т-лимфоцитов – Т-супрессоров (CD4+, CD25), подавляющих избыточную активность иммунных клеток. Они являются регуляторами иммунного ответа.
Патогенез
В результате мутации FOXP3 нарушается регуляция иммунного ответа, утрачивается аутотолерантность, происходят реакции иммунной аутоагрессии. Лимфоциты начинают атаковать собственные клетки, принимая их за чужеродные. В патологический процесс могут вовлекаться любые органы, наиболее часто страдают кожа, эндокринные железы и кишечник. Поражение поджелудочной и щитовидной железы приводит к сахарному диабету 1 типа и гипотиреозу.
При патоморфологическом исследовании кишечника обнаруживается атрофия ворсин с мононуклеарной инфильтрацией слизистой оболочки, некроз эпителиальных клеток, формирование крипт-абсцессов. Нередко возникает панцитопения, т.е. снижение в крови уровня всех форменных элементов – эритроцитов, лейкоцитов, тромбоцитов. Помимо аутоиммунных изменений ухудшается сопротивляемость бактериальным и вирусным инфекциям.
Классификация
Клинически заболевание не всегда протекает однотипно, что, вероятно, связано с различными мутациями FOXP3. Выделяют 2 вида IPEX-синдрома:
- Классическая форма. Характеризуется полной потерей функции белка скарфина, развернутой тяжелой симптоматикой и высокой летальностью на первом году жизни.
- Неполная форма. Благодаря остаточному функционированию скарфина наблюдается более мягкое моносимптомное течение с продолжительностью жизни от 20 до 30 лет.
Симптомы
Клиническая манифестация происходит практически сразу после рождения. Проявления могут быть крайне разнообразными, однако существует классическая триада признаков, которая присутствует у подавляющего числа больных – аутоиммунная энтеропатия (100% случаев), полиэндокринопатия (70-80% случаев), поражение кожных покровов (65-70% случаев).
Самым постоянным симптомом считается профузная секреторная диарея, из-за которой быстро нарастает белково-энергетическая недостаточность. Ребенок плохо прибавляет в весе, отстает в росте. В течение первых 6 месяцев дебютирует сахарный диабет – начинают беспокоить постоянная жажда, повышенное мочеотделение, мышечная слабость.
Диабету при IPEX-синдроме часто сопутствует гипотиреоз, из-за которого пациент становится вялым, сонливым и отечным, значительно отстает в нервно-психическом развитии. Высыпания по характеру похожи на экзематозные или псориатические. На коже появляются красные пятна, шелушащиеся бляшки, пузырьки, которые после разрыва оставляют язвочки.
Отличительная особенность кожных элементов – устойчивость к терапии антигистаминными и кортикостероидными препаратами местного действия. Вследствие аутоиммунной панцитопении у пациентов нередко наблюдаются бледность кожи и слизистых с желтушным оттенком, носовые и десневые кровотечения, спонтанное возникновение синяков. Возможны аутоиммунный гепатит, нефропатия, реактивное увеличение селезенки и периферических лимфатических узлов.
Осложнения
Для IPEX-синдрома характерно большое число осложнений. К наиболее частым относятся вторичные бактериальные или грибковые инфекции, присоединяющиеся к кожным высыпаниям. Из-за выраженного иммунодефицита ребенок подвержен и другим инфекционным заболеваниям (пневмонии, менингиту, туберкулезу), которые склонны к генерализации, порой – до септического состояния.
Другие неблагоприятные последствия отмечаются реже. Неонатальный сахарный диабет может дебютировать таким острым и неотложным состоянием как кетоацидоз или кетоацидотическая кома. Возможно массивное кровотечение и кишечная непроходимость. Аутоиммунная нефропатия у взрослых может привезти к хронической почечной недостаточности.
Диагностика
Курацией больных с IPEX-синдромом занимаются врачи-педиатры и генетики. Заподозрить заболевание помогает мужской пол ребенка, сочетание энтеропатии, эндокринопатии и кожных высыпаний. Для подтверждения диагноза назначается обследование, включающее:
- Общие лабораторные исследования. В общем анализе крови у многих пациентов отмечается снижение уровня гемоглобина, лейкоцитов, тромбоцитов, эритроцитов. Часто обнаруживается эозинофилия. В биохимическом анализе крови выявляется низкое содержание альбумина, электролитов, увеличение концентрации печеночных трансаминаз, глюкозы, гликированного гемоглобина. В анализе мочи возможна протеинурия.
- Иммунологические исследования. В крови определяется повышенный IgE, антитела к антигенам клеток кишечника (AIE-75, виллину), тиреопероксидазе, тиреоглобулину, панкреатическим бета-клеткам, глутаматдегидрогеназе, тирозинфосфатазе. В иммунограмме характерно снижение иммунорегуляторного индекса (CD4+/CD8+).
- Генетическое исследование. Основной анализ, позволяющий достоверно установить диагноз IPEX-синдрома. Методом полимеразной цепной реакции выявляется мутация гена FOXP3.
Дифференциальный диагноз проводится с наследственными первичными иммунодефицитами (синдром Вискотта-Олдрича, синдром Ди Джорджи), генетическими нарушениями обмена веществ, аутоиммунным полигландулярным синдромом. Изолированная аутоиммунная энтеропатия при моносимптомном течении требует дифференциальной диагностики с пищевой непереносимостью (целиакией), инфекционной диареей, гормонпродуцирующими опухолями (гастриномы, ВИПомы).
Лечение IPEX-синдрома
Пациенты подлежат обязательной госпитализации. На момент постановки диагноза из-за выраженной мальабсорбции возникает необходимость в парентеральном питании и инфузиях альбумина. Единственным эффективным методом лечения признана трансплантация стволовых клеток костного мозга. Ранняя ТГСК позволяет предотвратить развитие осложнений. Лекарственная терапия носит поддерживающий и симптоматический характер:
- Иммуносупрессоры. Для подавления аутоиммунного повреждения внутренних органов применяются глюкокортикостероиды (преднизолон), цитостатики (циклофосфамид, тамоксифен), антицитокиновые препараты (инфликсимаб). Эффективность перечисленных ЛС часто оказывается недостаточной.
- Гормональные средства. Для лечения сахарного диабета пожизненно назначаются инъекции инсулина. Подбор препаратов короткого и длительного действия осуществляется строго индивидуально. Для контроля эффективности лечения регулярно проводится мониторинг гликированного гемоглобина. При развитии гипотиреоза используется L-тироксин под контролем уровня ТТГ.
Прогноз и профилактика
IPEX-синдром является тяжелым заболеванием. При классической форме без своевременной диагностики и пересадки костного мозга смерть к концу первого года жизни наступает почти в 100% случаев. Основными причинами гибели выступают генерализованные вирусные и бактериальные инфекции. Описаны фатальные исходы, ассоциированные с вакцинацией.
Продолжительность жизни людей с неполным дефектом иммунной регуляции составляет 20-30 лет. Единственным методом профилактики считается пренатальная диагностика – молекулярно-генетическое исследование образца ворсин хориона. Анализ назначается в тех случаях, когда у близких родственников была обнаружена мутация FOXP3.
1. Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A Paradigm of Immunodefi ciency with Autoimmunity/ Barzaghi F, Passerini L, Bacchetta R.// Frontiers in Immunology – 2012 - №3.
2. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy/ Powell BR, Buist NRM, Stenzel P.// The Journal of Pediatrics – 1982 - №5.
3. Первичные дефекты иммунной системы, являющиеся следствием иммунной дисрегуляции/ Пашанов Е.Д., Румянцев А.Г. – 2008.
4. Х-сцепленные иммунная дисрегуляция, полиэндокринопатия и энтеропатия (IPEXсиндром): описание клинического случая и краткий обзор литературы/ Тихонович Ю.В., Петряйкина Е.Е., Рыбкина И.Г., Гаряева И.В., Тюльпаков А.Н.// Проблемы эндокринологии – 2014 – Т.60, №5.
IPEX-синдром: 8 советов родителям ребенка с редкой болезнью
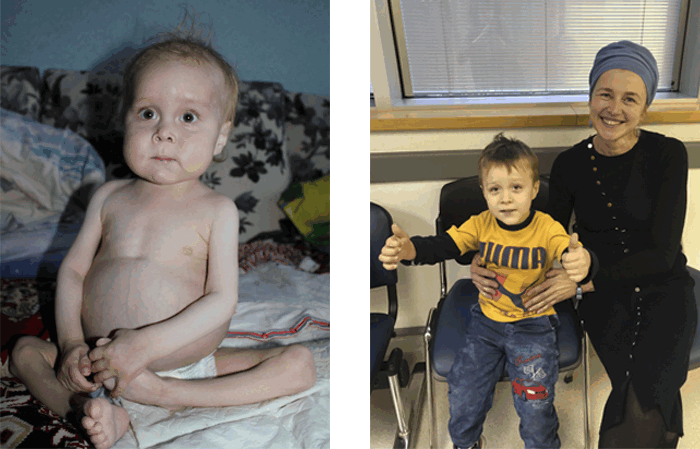
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Жизнь ребенка с IPEX-синдромом — это ежедневная борьба с редкой и опасной болезнью. После подтверждения диагноза, перед родителями встает множество задач по уходу и лечению. Однако главная цель — помочь детям принять свое заболевание и научить справляться с любыми трудностями. Ведь малышу так сложно понять, почему все так стремительно изменилось. Поэтому нередко маленький пациент отказывается выполнять рекомендации врачей. Избежать последствий подобных капризов возможно, если вовлечь малыша в процессы лечения. Мы подготовили для мам и пап список советов, которые сделают жизнь ребенка яркой и полноценной:
• Понимание болезни и лечения. Прежде всего родителям следует самим разобраться в во всех особенностях заболевания, методах лечения и возможных последствиях. В Хадассе для мамы и папы составляется полный перечень рекомендаций по образу жизни, питанию, физической активности и другим аспектам воспитания ребенка с IPEX-синдромом. Разговор с малышом о болезни должен быть выстроен в соответствие с его возрастом и уровнем развития (умственного и эмоционального):
— Младенцы (до 2 лет). Разлука с родителями, неприятные ощущения во время медицинских процедур, тревога и страх близких людей могут вызывать сильную эмоциональную реакцию у ребенка. Главная задача мамы и папы — создать комфортные условия дома и во время лечения. В Хадассе для маленьких пациентов выделен специальный корпус матери и ребёнка, в котором все помещения оборудованы в соответствии с нуждами детей. Родственникам разрешается находится с ребенком во время большинства процедур.
— Дошкольники (2-5 лет). Длительные госпитализации, постоянный прием лекарств и чрезмерное внимание врачей и родителей нередко становятся причинами капризов и истерик. Главная задача родителей — проявить терпение и твердость. Малышу необходимо в максимально доступной форме объяснить важность терапии и определенных ограничений.
— Дети младшего возраста (5-10 лет). Стремление к контролю над своей жизнью, чувство вины за болезнь, понимание явных отличий от сверстников часто приводят к отказу ребенка от лечения. В таком возрасте главная задача родителей — объяснить природу заболевания и дать ребенку возможность самостоятельно принимать некоторые решения. На данном этапе особенно важно хвалить малыша за любые успехи и соблюдение врачебных рекомендаций. Мама и папа могут в доступной игровой форме показать детям, как ухаживать за собой, принимать лекарства или отвечать на вопросы о своей болезни.
— Дети старшего школьного возраста (от 11 лет). Плохое самочувствие, переходный подростковый период, частые госпитализации и ограничения в жизни нарушают привычный уклад жизни ребенка. Вместо общения со сверстниками малыш вынужден длительное время находиться в больнице. Подобные перемены крайне негативно сказываются на эмоциональном состоянии и на социализации детей в обществе. Главная задача родителей вовлечь малыша в процесс лечения, установить между ним и врачами прочную связь и предоставить ему больше возможностей для контроля своего заболевания и жизни.
• Подготовка к медицинским процедурам. Родители должны понимать и уметь объяснить своим детям, что их ожидает в больнице, почему были назначены те или иные процедуры, кто будет их выполнять, будут ли они болезненными. Лечение IPEX-синдрома требует комплексного подхода и работы мультидисциплинарной команды врачей, включающей специалистов по орфанным болезням и трансплантации костного мозга. В Хадассе показатели успешного лечения при применении ТКМ превышают здесь все имеющиеся на сегодня стандарты. Пересадка костного мозга проводится под руководством профессора Полины Степенски — мирового эксперта по ТКМ, к которому приезжают пациенты со всего мира, в том числе из России.
• Возможности выбора. Когда малыши начинают осознавать свое состояние, у них нередко появляется чувство, что взрослые полностью контролируют их жизнь. Подобные эмоции часто приводят к скандалам, капризам и отказу от лечения. Избежать подобных конфликтных ситуаций возможно, предоставив ребенку право выбора, например, последовательности приема лекарств или одежды для больницы.
• Игровое воспитание. Ребенок с IPEX-синдромом испытывает различные сложности в социализации и общении с людьми. Родители могут помочь ему разобраться в своем заболевании с помощью игровых сценок. Например, во время развлекательных занятий мама и папа помогут подобрать слова, чтобы рассказать о своем состоянии друзьям. Подобные игры позволяют детям заранее подготовиться ко многим сложным вопросам и почувствовать себя увереннее.
• Подготовка сотрудников школы или детского сада. Жизнь ребенка с IPEX-синдромом будет отличаться от жизни его сверстников и одноклассников. По состоянию здоровья малышу может потребоваться срочная госпитализация или особые условия. Задача родителей обсудить с учителями и директором тактику поведения и общения, все возможные последствия болезни, ответить на вопросы о потенциальном влиянии IPEХ-синдрома на учебу (например, частые пропуски, усталость, ограничения физической активности).
• Контроль эмоций. Привычная жизнь навсегда меняется после подтверждения редкого и опасного заболевания. Негативные эмоции сказываются на состоянии здоровья ребенка и создают новые психологические проблемы, такие как:
- Нарушение настроения: чувство тревоги или страха, грусть и депрессия, безнадежность, раздражение, гнев, отсутствие интереса, чувство вины и беспокойство;
- Изменение поведения: перепады настроения, всплески агрессии, отказ от лечения или обследований и капризы;
- Проблемы с обучением: трудности с концентрацией, частые ошибки и снижение уровня успеваемости;
- Социальные нарушения: изоляция от сверстников и чувство одиночества;
- Семейные проблемы: напряженность в отношениях с близкими людьми, взаимные обвинения;
- Физические проблемы: изменения веса, нарушения сна, боли в животе или головные боли, усталость.
• Контроль здоровья. Родители — важная часть команды, сражающяся за жизнь и здоровье ребенка. Внимательность к симптомам, тщательный контроль и наблюдение за любыми изменениями помогут врачам назначить и скорректировать лечение. При первичном приеме у специалиста маме и папе необходимо иметь следующую информацию:
- Краткую историю болезни, написанную родителями или лечащим врачом;
- Копии исследований, подтверждающих диагноз;
- Хронологию важных событий — лечения, изменения в терапии и последующей реакции, операции;
- Список принимаемых лекарств;
- Список лекарств, на которые у малыша аллергия.
• Поиск положительных примеров. Пример ребенка, прошедшего успешное лечение, поможет настроиться на позитив и обрести уверенность. Среди маленьких пациентов Хадассы есть мальчик, который стал для многих из нас настоящим героем. История Ванечки Волохатюка и его мамы Ирины навсегда останется в сердце каждого сотрудника клиники. Долгие годы украинские врачи не могли поставить Ване верный диагноз. За несколько лет ребенок прошел десятки обследований, перенес 1 операцию и 14 наркозов. Только специалисты “Хадассы” выявили одно из редчайших в мире заболеваний — первичный иммунодефицит IPEX-синдром. 24 декабря 2015 года в “Хадассе” Ване была проведена трансплантация костного мозга (ТКМ). Недавние результаты обследований показали, что донорский мозг функционирует на 100%. Своими впечатлениями о лечении в Хадассе в своем видеоотзыве поделилась мама Ванечки — Ирина.
Автор: Редакция сайта
IPEX-синдром

Ваня с мамой во время одного из недавних визитов в клинику «Хадасса» (2020 год)
В медицинском центре «Хадасса» очень успешно лечат как IPEX-синдром, так и другие виды первичных иммунодефицитов (ПИД). Один из наиболее известных случаев излечения ребенка с IPEX в этой клинике — история Вани Волохатюка (Украина).
В течение 2 лет мальчику не могли поставить правильный диагноз, это удалось только генетикам клиники «Хадасса». Затем известный трансплантолог проф. Полина Степенски сделала маленькому пациенту ТКМ. Ваня сразу же пошел на поправку, сегодня это совершенно здоровый ребенок.
«Хадасса» по праву гордится одним из самых высоких уровней полного излечения детей с ПИД в мире — при применении аллогенной трансплантации костного мозга он составляет 94%.
Что такое IPEX-синдром

Проф. Полина Степенски
Руководитель отделения ТКМ и иммунотерапии у детей и взрослых клиники «Хадасса»
Одним из редких врожденных аутоиммунных расстройств, состоящих в группе первичных иммунодефицитов, является IPEX-синдром. Это тяжелое х-сцепленное заболевание, вызванное мутациями гена FOXP3. Синдром IPEX проявляется аутоиммуногенными реакциями и характеризуется инфекциями, эндокринными нарушениями, диареей, кожными поражениями. Статистические данные о частоте встречаемости IPEX-синдрома отсутствуют. В настоящее время насчитывается около 140 пациентов с подтвержденным диагнозом. Но специалисты считают, что численность случаев недооценена в силу редкости болезни. IPEX-синдрому характерна ранняя манифестация, в течение 3-6 месяцев после рождения ребенка. Нередко дебют заболевания наблюдается в первые дни и недели жизни. IPEX-синдрому подвержены в основном мальчики.
IPEX-синдром или Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии и энтеропатии (от англ. immunodysregulation polyendocrinopathy enteropathy X-linked syndrome) был впервые описан в 1982 году. Это наследственное, редкое заболевание, которое сопровождается дисфункцией эндокринных желез, воспалением кишечника и персистирующей белоктеряющей диареей, а также поражениями слизистых оболочек кожи. IPEX-синдром как и неонатальный сахарный диабет, вызван мутациями гена FOXP3, который отвечает за развитие и функцию регуляторных Т-лимфоцитов. В результате генного дефекта развиваются аутоиммунные поражения органов и систем. Дисфункция регуляторных Т-клеток приводит к недостаточности антибактериального и противовирусного иммунитета, поэтому у детей с IPEX-синдромом наблюдаются тяжелые инфекционные болезни. Тяжесть патологии и клинические признаки могут отличаться. При отсутствии эффективного лечения IPEX-синдрома, летальный исход неизбежен в течение 1-2 лет.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
МКБ-10. Согласно международной классификации болезней 10 пересмотра IPEX-синдром входит в группу аутоиммунных полиэндокринных синдромов и имеет код E31.0.
Причины
IPEX-синдром обусловлен мутацией гена FOXP3, который расположен в локусе Хр11.23 Х-хромосомы. Его функция — кодировать белок, необходимый для созревания Т-регуляторных (TR) лимфоцитов. Неполноценность TR-клеток на фоне мутации приводит к развитию аутоиммунных расстройств. Наследуется IPEX-синдром по сцепленному с Х-хромосомой рецессивному признаку. Поэтому болезни подвержены мальчики. Поскольку у женщин 2 Х-хромосомы, одна из них остается с нормальным геном. Женщины являются носителями мутантного гена и могут передавать его своим сыновьям, будучи здоровыми.
Симптомы

Проф. Орли Эльпелег
Заведующая отделением генетических и метаболических заболеваний
IPEX-синдром проявляется триадой клинических признаков: поражениями кожи, аутоиммунными расстройствами, энтеропатией. Заболевание у детей манифестирует в первые недели или месяцы жизни. При IPEX-синдроме наблюдаются следующие симптомы: ониходистрофия, алопеция, ногтевые повреждения, множественные кожные высыпания (экзема, псориаз, различные виды дерматита), явления гиперкератоза, шелушения кожи. Развивается инсулинозависимый сахарный диабет 1 типа, а также хроническое воспаление щитовидной железы, приводящее к гипертиреозу.
У детей с IPEX-синдромом снижается аппетит и прибавка в весе, они значительно отстают в росте. Часто болезнь сопровождается труднокупируемой лихорадкой, секреторной стойкой диареей, анемией, вздутием живота, нарушением усвоения основных питательных веществ, рвотой, кишечной непроходимостью, колитом, гастритом. Нередко при IPEX-синдроме отмечается спленомегалия, гиперплазия лимфоузлов (лимфаденопатия), увеличение аденоидов и миндалин.
Кроме того, у маленьких пациентов выявляют дефицит различных видов кровяных клеток, что вызывает повышенную кровоточивость и ухудшает свертываемость крови. Дети с IPEX-синдромом страдают от частых инфекций локального и системного характера. Аутоиммунные расстройства приводят к миозиту, пневмонии, дисфункции печени и почек.
Диагностика
Верификация диагноза проводится на основе анамнестических данных, изучения семейной истории болезни, физикального осмотра, лабораторных исследований. Общий и биохимический анализ крови позволяет оценить показатели иммунного статуса, выявить аутоиммунную энтеропатию, сахарный диабет, цитопению, тиреоидит, то есть обнаружить аутоантитела против инсулина, тироглобулина, энтероцитов, тромбоцитов и т.д. Для постановки окончательного диагноза IPEX-синдром проводят молекулярно-генетическую диагностику на мутации в гене FOXP3. Чтобы оценить общее состояние пациента требуется КТ брюшной полости, ФГДС с гистологией, ЭКГ, а также УЗИ, МРТ и эндоскопическое исследование пораженных органов и структур.
IPEX-синдром дифференцируют с синдромом Оменна и Вискотта-Олдрича, промежуточными формами комбинированного иммунодефицита, переходным диабетом новорожденных, гипоплазией поджелудочной железы и другими заболеваниями, схожими по симптоматике.
Лечение
IPEX-синдром полностью не излечим, врачи применяют методы поддерживающей терапии, которые позволяют минимизировать проявления болезни, избежать осложнений и продлить жизнь. Для коррекции различных патологических состояний врачи используют глюкокортикостероиды, противовирусные и антибиотические средства, препараты моноклональных антител.
Детям с IPEX-синдромом при тяжелой энтеропатии необходима нутритивная поддержка — искусственное энтеральное питание, подразумевающее введение в организм питательных веществ, витаминов, минералов. Также в процессе поддерживающей терапии при IPEX-синдроме детям требуется инсулинотерапия, наружное лечение кожных поражений, купирование диспептических явлений, внутривенное введение иммуноглобулинов, коррекция других патологических состояний.
В случае неэффективности заместительной и иммуносупрессивной терапии специалисты при IPEX-синдроме рассматривают пересадку костного мозга. Процедуру необходимо выполнить как можно раньше, до 3,5-4 месяцев после рождения, пока не возникло серьезных поражений органов-мишеней. Трансплантация гемопоэтических стволовых клеток от совместимого донора способствует восстановлению иммунной системы и нормальной функции TR-клеток.
Осложнения
Тяжелая форма IPEX-синдрома нередко приводит к септическим осложнениям, рецидивирующим эпизодам кишечной непроходимости, частым пневмониям, генерализированным инфекциям, гепатиту, дисфункции печени, селезенки и почек. Прогрессирование неонатального сахарного диабета может привести к тяжелой дегидратации, ацидозу.
Прогноз заболевания и профилактика
Первичная профилактика IPEX-синдрома невозможна, поскольку это врожденное заболевание. Будущим родителям с наличием генного дефекта рекомендована консультация генетика. Прогностическая картина во многом зависит от тяжести симптоматической картины, поскольку проявления IPEX-синдрома вариабельны. Классическая форма заболевания характеризуется неблагоприятным прогнозом, дети погибают в течение 1 года жизни. При «мягких» мутациях, а также эффективной иммуносупрессивной терапии и пересадке костного мозга, дети доживают до 2-3 декады жизни. В случае своевременной диагностики и адекватного лечения IPEX-синдрома складывается благоприятная прогностическая картина.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Читайте также: