Синдром Карпентера у плода
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Синдром Карпентера принадлежит к группе редких генетических расстройств. При данном заболевании кости черепа как бы смешаются, и черепа головы выглядит «угловатым» (акроцефалия), также при этом возникает синдактилия (сращение) некоторых пальцев рук и ног, и может быть полидактилия (вместе 5 пальцев у человека 6 больше пальцев).
Этот синдром чаще всего обнаруживается сразу после рождения. При этом заболевании макушка головы напоминает конус (акроцефалия), а также выглядит укороченной и широкой. Также при данном заболевании может возникать ассиметрия головы и лица (краниофациальная ассиметрия). При данном заболевании могут возникать другие патологии лица и черепа, которые различны у каждого пациента.
Обычно у людей с данным заболеванием патологически короткие пальцы рук и ног, частичная фузия мягких тканей между определенными пальцами, а также наличие лишних пальцев рук или ног (полидактилия). Вдобавок к этому, у человека могут быть другие патологии, такие как очень низкий рост, врожденные патологии сердца, легкая или средняя степень ожирения. У мужчин при данном заболевании могут не функционировать яички. Также у многих людей с данным заболеванием наблюдается легкая или тяжелая степень умственной отсталости, однако, в большинстве случае, ум у пациента в порядке. Это заболевание передается генетически по аутосомно-рецессивному типу.
Молекулярно-генетической причиной заболевания являются мутации в гене RAB23, кодирующем белок, участвующий в важной негативной регуляции работы продукта гена SHH, определяющего развитие клеток на различных этапах эмбриогенеза.
Ген RAB23 кодирует белок, участвующий в важной негативной регуляции работы продукта гена SHH, определяющего развитие клеток на различных этапах эмбриогенеза.
Заболевание характеризуется акроцефалией (высокий “башенный” череп), краниосиностозом (преждевременным зарастанием черепных швов, ограничивающим рост черепа и приводящим к его деформации), ожирение.
На ногах выявляют преаксиальную полидактилию (увеличение количества пальцев с I по IV), кожную синдактилию (полное или частичное сращение соседних пальцев только мягкими тканями). На руках часто выявляют постаксиальную полидактилию (дополнительный мизинец), широкий или расщепленный большой палец, мягкотканную синдактилию, отсутствие средних фаланг.
Обычно у людей с данным заболеванием патологически короткие пальцы рук и ног, могут быть другие патологии, такие как очень низкий рост, врожденные патологии сердца, легкая или средняя степень ожирения. У мужчин при данном заболевании могут не функционировать яички. Также у многих людей с данным заболеванием наблюдается легкая или тяжелая степень умственной отсталости, однако, в большинстве случае, интеллект у пациента в порядке.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
Синдром Карпентера ( Акроцефалополисиндактилия 2 типа )
Синдром Карпентера (акроцефалополисиндактилия 2-го типа) — это чрезвычайно редкое наследственное заболевание, обусловленное мутацией гена RAB23. Патология передается по аутосомно-рецессивному типу. Болезнь имеет характерную триаду симптомов: раннее заращение черепных швов (краниосиностоз) с врожденными дизморфиями лицевого скелета, множественные пороки развития конечностей, ожирение. Диагностика синдрома Карпентера включает рентгенографию или КТ скелета, эхокардиографию, молекулярно-генетическую диагностику для выявления специфической мутации. Основу лечения составляет хирургическая коррекция краниосиностоза, создающая условия для развития головного мозга.
МКБ-10
Общие сведения
Заболевание впервые описано английским педиатром Джорджем Альфредом Карпентером, в 1909 году заметившим характерный набор симптомов у 3-х детей из одной семьи. Генетические предпосылки появления патологии были установлены в 2000 г. Болезнь встречается крайне редко, в литературе описано всего 40 случаев, ввиду чего статистические показатели заболеваемости отсутствуют. Несмотря на редкость синдрома, он не теряет своей актуальности в современной генетике, поскольку требует раннего выявления, проведения комплекса хирургических мероприятий в младенческом возрасте.
Причины
Синдром возникает вследствие генного дефекта RAB23, кодирующего одноименный белок. Ген расположен на коротком плече 6 хромосомы, состоит из 237 аминокислотных остатков. Для развития заболевания необходимо, чтобы ребенок получил по одной копии мутантного гена от каждого родителя. При этом вероятность синдрома Карпентера составляет 25%, независимо от пола новорожденного.
Патогенез
По биохимической структуре RAB23 представляет собой гуанозинтрифосфатазу суперсемейства Ras. Основная функция протеина — участие во внутриклеточном транспорте белков, что необходимо в процессе закладки и дифференцировки тканей. Он располагается в клеточных мембранах, цитоплазме клеток, везикулах и эндосомах. Кроме того, RAB23 контролирует активность гена SHH, отвечающего за развитие клеток в периоде онтогенеза.
При дефиците протеина вследствие синдрома Карпентера в первую очередь страдают процессы дифференцировки костной ткани. В результате этого происходит раннее срастание черепных швов, что формирует замкнутое пространство головы без возможности его увеличения соответственно росту головного мозга. Также патология RAB23 вызывает нарушения сигнальных каскадов при формировании центральной нервной системы.
С учетом особенностей зарастания швов формируется 2 типа краниосиностоза — сагиттальный (длинный узкий череп с выступающим лбом и большим затылком) и бикорональный (широкий короткий череп с деформированными лбом и глазницами). Тип краниосиностоза оказывает наибольшее влияние на особенности формирования неврологических расстройств.
Симптомы
Костные аномалии при болезни Карпентера формируются во внутриутробном периоде, поэтому они определяются сразу после рождения. Родители обращают внимание на вытянутую башнеобразную форму головы (акроцефалию). Со временем череп деформируется из-за раннего закрытия швов (краниосиностоза), которые ограничивают рост костей и не позволяют голове увеличиваться в объеме. Также наблюдаются множественные врожденные аномалии лицевого скелета.
Краниосиностоз проявляется разнообразными симптомами. При бикорональной деформации черепа отмечаются патологии зрительного аппарата: косоглазие, слезотечение, экзофтальм (выпячивание глазных яблок). Характерна задержка психомоторного развития: ребенок поздно начинает держать голову, плохо сидит и стоит, с трудом учится ходить. Также большое значение имеет косметический дефект, вызывающий у пациента при отсутствии коррекции психологическую травму.
Еще один типичный признак синдрома Карпентера — аномалии развития пальцев рук и ног. Зачастую возникает кожная синдактилия — сращение мягких тканей соседних пальцев, также возможно отсутствие средних фаланг, деформация большого пальца. Некоторые больные имеют шестипалость, причем на руках чаще есть добавочный мизинец, а на ногах — любой палец с I по IV. По мере взросления пациента становится заметна короткопалость, отставание в росте.
Дизэмбриогенез внутренних органов, как правило, проявляется врожденными сердечными аномалиями, которые выявляются у трети больных. Постепенно у детей развивается ожирение, сначала легкой, а затем средней степени, с преимущественным распределением жира по лицу, телу, проксимальным частям конечностей. У мальчиков возможен крипторхизм. Интеллектуальный уровень примерно у 75% пациентов соответствует легкой умственной отсталости.
Осложнения
Наиболее серьезная проблема при краниосиностозе — внутричерепная гипертензия, которая проявляется срыгиваниями, повторной рвотой, беспокойством вследствие сильной головной боли. Возможно развитие судорог, менингеального синдрома, в тяжелых случаях наступает кома. Опасность представляют пороки сердца, чреватые критическими нарушениями кровообращения.
Диагностика
Обследованием пациента занимается неонатолог /педиатр, по показаниям к диагностике подключают челюстно-лицевого хирурга и нейрохирурга, генетика, детского кардиолога. Заподозрить синдром Карпентера удается по характерным фенотипическим особенностям, однако для подтверждения диагноза проводятся лабораторные и инструментальные исследования:
- КТ черепа. Для уточнения степени костных деформаций, состояния черепных швов применяется спиральная компьютерная томография, которая дает максимально детальное изображение всех структур, помогает в выборе тактики хирургического лечения.
- УЗИ сердца. Учитывая высокую вероятность кардиологических пороков, всем пациентам с подозрением на болезнь Карпентера показана эхокардиография. При выявлении аномалий дополнительно делают ЭКГ, МРТ сердца, катетеризацию сердца.
- Генетическое тестирование. Секвенирование генома с целью диагностики мутантного гена RAB23 — единственно возможный способ 100% подтвердить диагноз. После установления заболевания целесообразно провести генетическое исследование родителям, ближайшим кровным родственникам.
- Пренатальная диагностика. При отягощенной наследственности или обнаружении подозрительных признаков при УЗИ-скрининге выполняется биопсия хориона во 2-м триместре беременности. Полученный материал используется для молекулярно-генетического тестирования.
Лечение синдрома Карпентера
Консервативная терапия
Этиопатогенетическое лечение заболевания не разработано. Чтобы корректировать избыточный вес тела, больным рекомендуется соблюдение пожизненной диеты со сниженным калоражем. Для уменьшения выраженности возможных сопутствующих расстройств показаны:
- Специальные методы обучения. Детям с умственной отсталостью необходимо обучаться по специальным программам, составленным олигофренопедагогами. Также важную роль играет выработка навыков бытового самообслуживания.
- Коррекция эмоциональных расстройств. При наличии признаков невротических или депрессивных нарушений используются методы психотерапии, подбираются успокоительные фитопрепараты, мягкие антидепрессанты.
- Офтальмологическая помощь. При снижении остроты зрения, косоглазии, слезотечении и других нарушениях зрения, типичных для бикоронального кранисиностоза, больному может потребоваться соответствующая коррекция.
Хирургическое лечение
При синдроме Карпентера основной задачей становится коррекция краниосиностоза, выполняемая только хирургически. Оптимальный возраст для проведения операции — 3-9 месяцев. Раннее устранение деформации черепа создает благоприятные условия для физического и интеллектуального развития пациента. Нейрохирурги делают эндоскопическую или открытую краниопластику, используют биодеградируемые и артифицированные титановые имплантаты.
Больным с синдактилией рекомендована операция по разделению пальцев. При терминальном типе сращения хирургическое вмешательство показано во втором полугодии жизни младенца, в остальных случаях операция проводится в возрасте 4-5 лет. При шестипалости применяется метод ампутации добавочного пальца. В случае врожденных сердечных пороков требуется консультация кардиохирургов для индивидуального подбора способа устранения дефектов.
Прогноз и профилактика
При своевременном выявлении синдрома Карпентера и проведении хирургической коррекции краниосиностоза удается полностью нивелировать признаки заболевания. Прогноз благоприятный при условии отсутствия умственной отсталости и тяжелых пороков сердца. Профилактика патологии предполагает медико-генетическое консультирование родителей, у которых уже есть больной ребенок или отягощен семейный анамнез.
1. Краниосиностоз. Опыт лечения на базе одной клиники/ А.А. Жайлганов// Нейрохирургия и неврология Казахстана. — 2020. — №1.
2. Сочетание синдрома Карпентера с эпилепсией. Клинический случай/ С.С. Стецура// VI Балтийский конгресс по детской неврологии. — 2016.
3. Carpenter syndrome/ Hidestrand, Pip, Henry Vasconez, and Carol Cottrill// Journal of Craniofacial Surgery. — 2009. — №1.
Пренатальная УЗИ черепно-лицевых аномалий плода
Пренатальная диагностика черепно-лицевых аномалий является важным и обязательным аспектом, так как аномалии этих структур могут указывать на наличие других, более тонких аномалий, синдромов, хромосомных мутаций или даже более редких состояний, таких как инфекции или нарушения обмена веществ.
Она остается сложным процессом, особенно в первом триместре. Частота выявления черепно-лицевых аномалий(ЧЛА) варьируется в зависимости от типа аномалии, ее тяжести, гестационного возраста, сопутствующих аномалий, а также методов и технологий ультразвуковых исследований. Так распространенность хейлосхизиса и краниосиностоза составляет около 0,15% и 0,05% соответственно.
Различные профессиональные общества, в том числе Международное общество ультразвуковой диагностики в акушерстве и гинекологии (ISUOG), Американский институт ультразвука в медицине и Федерация акушерства и гинекологии Азии и Океании, выпустили рекомендации по обследованию лица и черепа во втором триместре беременности. Основная цель этой статьи – предоставить обновленную информацию о пренатальном выявлении ЧЛА с целью повышения точности диагностики.
Изменения мозгового отдела черепа
Размер, форму, целостность и плотность кости черепа можно оценить, когда измеряется размер головы и когда исследуются структуры головного мозга. Череп имеет овальную форму и непрерывную эхогенную структуру, прерываемую только узкими эхолуцентрическими швами. Аномальные особенности (Рис. 1-3) и связанные с ними аномалии показаны в Таблице 1.
Рисунок 1: Плод во втором триместре с тригоноцефалией.
Аксиальный вид головки плода (H) показывает лоб треугольной формы (стрелка).
Рисунок 2: Плод во втором триместре с брахицефалией. Аксиальный вид головы плода (H) показывает, что форма черепа короче, чем типичная (стрелка).
Рисунок 3: Плод во втором триместре со скафоцефалией.
Аксиальный вид головки плода (H) показывает длинную (указатель) и узкую голову плода (стрелки).
| Критерий | Показатель | Аномалия |
| Размер | Малый | Микроцефалия |
| Большой | Макроцефалия | |
| Форма | Не овальная, лимоноподобная | Расщелина позвоночника, трисомия 18 хромосомы или дисплазия скелета |
| Целостность | Дефект в кости черепа с выпячиванием мозговой ткани | Энцефалоцеле |
| Плотность | Отсутствие эхогенности, череп легко сдавливается | Слабая минерализация, несовершенный остеогенез или гипофосфатазия |
Диагностика краниосиностоза выражается в потере гипоэхогенности в сегменте основных швов черепа, вместе с расширением других ортогональных швов. Косвенные признаки, включая аномальный цефальный индекс (ЦИ), форму черепа (Таблица 2) и/или морфологию лица, такую как гипотелоризм или гипертелоризм, могут предшествовать закрытию швов на 4-16 неделе. ЦИ ниже 70% или выше 85% указывают на долихоцефалию и брахицефалию соответственно.
Измерение окружности имеет роль также. Так как отклонение окружности головы более чем на 3 стандартных единицы ниже или на 2 выше среднего значения, в зависимости от гестационного возраста, являются подсказкой для возможного диагноза микроцефалии или макроцефалии, соответственно.
Однако использование этих эталонных значений может привести к чрезмерной диагностике микроцефалии. Следует обращать внимание на другие поддерживающие признаки, которые включают деформацию лба, плоский затылок или внутричерепное содержимое, которое является ненормальным или невидимым.
ПРАВИЛЬНО ЛИ ВЫ УХАЖИВАЕТЕ ЗА УЗ-АППАРАТОМ?
Скачайте руководство по уходу прямо сейчас
Изменения лицевого отдела черепа
Оценка лицевого отдела черепа должна проходить в трех плоскостях для оценки различных лицевых структур, поскольку это облегчает обнаружение аномалий (таблица 3). Согласно руководству ISUOG, минимальная оценка лица плода включает наличие обеих орбит, оценку носа/ноздрей, наличие рта и, предпочтительно, оценку профиля лица и губ.
| Плоскость | Структура | Показатель | Аномалия |
| Венечная | Губы | Нарушение целостности | Хейлосхизис |
| Рот | Маленький или непрерывный, открытый | Микростомия или синдромы | |
| Нос | Сплющенный или в наличии одна ноздря | Гипоплазия или синдром одной ноздри | |
| Пальпебральная борозда | Наклон вверх или вниз | ||
| Поперечная | Орбиты | Малые, отсутствуют, аномальный межокулярный диаметр | Микрофтальмия / анофтальмия, гипотелоризм / гипертелоризм |
| Медиальная киста | Киста слезного мешка | ||
| Хрусталик | Эхогенный | Катаракта | |
| Зубные почки | Расщелины, ненормальное число | Волчья пасть, олигодонтия / анодонтия | |
| Нижняя челюсть | Маленькая | Микрогнатия | |
| Язычок | Отсутствует или двойной “знак равенства” | Расщелина язычка | |
| Уши | Неправильный размер, форма, местоположение или развернуты | Аномалия развития уха | |
| Сагиттальная | Лоб | Выпуклый | Скелетная дисплазия |
| Сплющенный | Микроцефалия | ||
| Нависший | Добавочный нос | ||
| Нос | Сплющенный | Синдромы | |
| Отсутствие или укорочение носовой кости | Анеуплоидия | ||
| Верхняя челюсть | Премаксиллярное выпячивание | Двусторонняя расщелина лица | |
| Мягкое небо | Нет мягкого неба или «знак равенства» | Расщелина мягкого неба, добавочный язычок | |
| Губной желобок | Длинный или короткий | Синдромы | |
| Нижняя челюсть | Маленькая | Микрогнатия | |
| Симметричность | Асимметрия лица | ||
| Язык | Большой, выпячивается, массово смещен назад | Макроглоссия, глоссоптоз опухоли | |
| Уши | Ненормальный размер, форма или местоположение или развернуты | Аномалия развития уха |
Расщелина диагностируется при потере целостности губы с одной или обеих сторон на венечной проекции (рис. 4, 5). Двусторонняя расщелина губы предполагает наличие предчелюстной выпуклости в сагиттальном виде (рис. 6). Трудно диагностировать неполную расщелину губы (рис. 7), зачастую видна лишь расщелина неба. Используя цветной допплер, можно увидеть поток околоплодных вод, в норме проходящий через ноздри во время дыхательной деятельности, в случае аномалии через небо, когда у него есть расщелина.
Рисунок 4: Плод второго триместра с односторонней расщелиной губы.
Венечный вид лица плода показывает потерю целостности (стрелка) верхней губы (L).
Рисунок 5: Плод второго триместра с двусторонней расщелиной губы.
Венечный вид лица плода показывает потерю целостности (стрелки) верхней губы (L) с обеих сторон (1 и 2).
Рисунок 6: Плод второго триместра с предчелюстной выпуклостью.
Сагиттальный вид лица плода показывает массу мягких тканей (стрелка), выступающую вперед под носом (N).
Рисунок 7: Плод второго триместра с частичной односторонней расщелиной губы. Венечная плоскость лица плода показывает частичную потерю целостности (стрелки) верхней губы (L).
Профиль лица можно оценить по срединно сагиттальной проекции. В частности, могут быть обнаружены лобовые выпуклости (рис. 8), микрогнатия (рис. 9) или плоский нос (рис. 10). С латеральной стороны можно оценить аномалии уха (рис. 11). Оба глаза и их аномалии могут быть оценены в осевой плоскости (рис. 12-14).
Рисунок 8: Плод во втором триместре с выпуклым лбом.
Среднесагиттальный вид показывает прямую выпуклость (стрелка) лба (FH).
Рисунок 9: Плод во втором триместре с микрогнатией.
Среднесагиттальный вид показывает маленький и отступающий (стрелка) подбородок (С).
Рисунок 10: Плод во втором триместре с плоским носом.
Среднесагиттальный вид показывает плоский (стрелка) нос (N).
Рисунок 11: Плод во втором триместре с аномалией уха.
Боковой сагиттальный вид показывает маленькое (стрелки) правое (R) ухо (E) с потерей нормального строения.
Рисунок 12: Плод во втором триместре с гипотелоризмом.
Аксиальный вид показывает аномально уменьшенное расстояние (стрелки) между двумя орбитами (круг).
Рисунок 13: Плод во втором триместре с двусторонней катарактой.
Аксиальный вид показывает эхогенность (стрелки) в хрусталике (L) обоих глаз.
Рисунок 14: Плод во втором триместре с выпячиванием между двумя орбитами. Аксиальный вид показывает массу мягких тканей (стрелки), выступающую между двумя орбитами (кружками).
Стоит отличать микрогнатию (маленькая нижняя челюсть) и ретрогнатию – смещенние челюсть к зади. Дифференцируется с помощью определения нижнего угла лица в отношении ширины нижней челюсти к ширине верхней челюсти. При отягощенном наследственном анамнезе или при подозрении на аномалию, могут быть выполнены измерения структур плода, таких как длина костей носа, длина уха, длина верхней челюсти, а также диаметр глаза и межглазного пространства.
3D/4D объемная визуализация
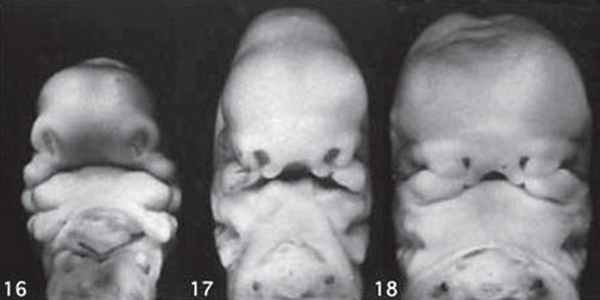
Использование трехмерного ультразвука, включая визуализацию поверхности, многоплоскостное и многослойное изображение, позволяет точно оценить различные черепно-лицевые структуры и их аномалии, включая расщелину неба (рис. 15, 16) и краниосиностоз. Изображения могут быть полезны для консультирования (рис. 17, 18). Использование 3D (рис. 19) может помочь дифференцировать закрытые и открытые швы. Использование 4D может быть использовано для оценки выражений лица.
Рисунок 15: Плод второго триместра с двусторонней расщелиной губы и неба. 3D аксиальный вид показывает потерю целостности (стрелки) губы (L) и неба (P) с обеих сторон.
Рисунок 16: Плод второго триместра с расщелиной губы и неба по средней линии. 3D аксиальный вид показывает потерю целостности (стрелки) губы (L) и неба (P) по средней линии. Цветные рамки справа показывают ориентацию активных изображений слева: аксиальный вид (1), сагиттальный вид (2) и венечный вид (3).
Рисунок 17: Плод второго триместра с односторонней расщелиной губы.
3D поверхностное изображение лица плода показывает расщелину (стрелка) на верхней губе (L).
Рисунок 18: Плод второго триместра с расщелиной средней линии.
3D поверхностное изображение лица плода показывает расщелину (стрелка) по средней линии верхней губы (L).
Рисунок 19: Плод во втором триместре с краниосиностозом.
3D поверхностное изображение в скелетном режиме, аксиального вида черепа показывает сужение (стрелки) венечного шва (CS) и части переднего родничка (AF).
УЗИ в первом триместре
В период между 11 и 13 неделями, в течении 6-ти дней беременности, рекомендации ISUOG предлагают измерять диаметр бипариетального отдела и окружность головы, а также оценивать целостность и эхогенность черепа. Можно попытаться оценить орбиты, межорбитальные расстояния, профиль лица, уши и целостность рта и губ. В первом триместре можно выявить уплощение профиля (рис. 20). Однако некоторые черепно-лицевые аномалии, такие как краниосиностоз, не могут быть диагностированы в первом триместре, и, таким образом, сканирование аномалии во втором триместре остается стандартом анатомической оценки плода.
Рисунок 20: Плод первого триместра с плоским профилем лица.
Сагиттальный вид показывает плоский лоб (FH) (стрелка), нос (N) (стрелка) и опускающийся подбородок (C) (изогнутая стрелка).
Подальшие исследования
При обнаружении расщелины неба важно определить, является ли она односторонней, двухсторонней или срединной, а также имеется ли какая-либо расщелина пластины или амниотическая полоса, потому что с этим варьируется прогноз и связанные с ним условия. Комбинированная расщелина губы и неба встречается чаще, чем сама расщепленная губа, и связанные с этим проблемы являются более серьезными. Односторонние / двусторонние и срединные расщелины губы считаются различными состояниями, поскольку у них разное эмбриологическое происхождение.
Всякий раз, когда обнаруживается черепно-лицевая аномалия, важно выполнить детальное сканирование для поиска дополнительных аномалий, особенно других потенциально тонких пороков развития лица, центральной нервной системы, сердца или конечностей. Многие черепно-лицевые аномалии, в том числе расщелины лица, микрогнатия, краниосиностоз, гипертелоризм / гипотелоризм, микрофтальмия / анофтальмия, катаракта и анотия / микротия, связаны с различными синдромами и состояниями (таблица 4).
В целом, 10% расщелин сопровождались хромосомной аномалией и 27% имеют ассоциированные аномалии. Примерно 15% случаев краниосиностоза являются синдромальными. Выпячивание языка может быть признаком синдрома Беквита-Видемана или синдрома Дауна.
| Аномалия | Сопутствующие пороки | Синдромы |
| Расщелина лица | Верхние конечности | Эктродактилия, эктодермальная дисплазия, синдром расщепления, орально-лицевой синдром I типа |
| Расщелина лица | Другие аномалии лицевого черепа и сердца | Синдром CHARGE (колобома, аномалия сердца, атрезия хоан, умственная отсталость, аномалии половых органов и ушей) |
| Микрогнатия | Уши | Синдром Голденхара |
| Микрогнатия | Конечности | Синдром гипогенеза рта-нижней челюсти-конечностей, синдром Нагера, синдром расщепления, синдром Робертса |
| Микрогнатия | Другие аномалии лицевого черепа | Синдром Робена, Синдром Тричера Коллинза |
| Краниосиностоз | Другие аномалии лицевого черепа и конечностей | Синдром Аперта, синдром Крузона, синдром Карпентера, танатофорная дисплазия, синдром Пфайффера, синдром Сэтре-Чотзена, синдром Мюнке, синдром Джексона-Вейсса, синдром Антли-Бикстлера, синдром Вольфа-Гиршхорна (4р) |
Прогноз
Прогноз и ведение зависят от типа и тяжести черепно-лицевых аномалий, основного состояния или хромосомных мутаций. Если прогноз плохой, как, например, в случае множественных аномалий или связанных с ними анеуплоидий, может быть предложен вариант прерывания беременности в зависимости от гестационного возраста и местных норм. Альтернативно, продолжение беременности с пренатальным консультированием подходит для легких и изолированных нарушений, таких как заячья губа.
Изолированная макроцефалия (с окружностью головы, которая на 2-3 стандартных отклонения выше среднего значения для гестационного возраста), долихоцефалия и брахицефалия обычно имеют благоприятный исход.
В нашем каталоге вы можете выбрать аппараты для исследований в области акушерства и гинекологии от передовых производителей ультразвукового оборудования. Свяжитесь с нашим менеджером, для уточнения деталей и выбора оптимальной для вас модели.
Новый эхографический признак для изучения нижней челюсти плода в норме и при патологии (микрогнатии) в I триместре беременности (11-14 недель)
Московский областной НИИ акушерства и гинекологии, Москва.
Кафедра медицинской генетики, курс пренатальной диагностики РМАПО, Москва.
Clinica lаs Condes, Santiago, Chili.
Важнейшим маркером генетических синдромов как хромосомного, так и нехромосомного генеза, является микрогнатия. Микрогнатия (нижняя микрогнатия, микрогения) - аномалия развития нижней челюсти, характеризующаяся ее гипоплазией. Диагностика этого состояния при трисомии 18 и триплоидии доходит до 80% [1, 2]. При введении в поисковую систему OMIM термина "micrognatia" можно встретить 447 различных синдромов и ассоциаций, в синдромальное ядро которых входит этот важный генетический маркер. Одна из самых крупных работ в мире по изучению этого маркера принадлежит D. Paladini и соавт. [3], которые описали более 50 случаев микрогнатии в сочетании как с хромосомными [4], так и нехромосомными синдромами и ассоциациями. Степени микрогнатии рассматривались от крайней - агнатии, входящей в состав аутосомно-рецессивного синдрома агнатии, голопрозэнцефалии (отоцефалии) [5, 6]. Отоцефалия - чрезвычайно редкая аномалия, при которой встречаются грубые лицевые дизморфии: недоразвитие или тяжелая гипоплазия нижней челюсти, неправильное положение ушей (рис. 1, 2), которые могут быть объединены и чаще всего располагаются на шее плода [7]. Также крайне выраженная степень микрогнатии может встречаться при окуло-ауриколофронтоназальном синдроме. Он был выделен в самостоятельную нозологическую группу, объединяющую симптомы как фронтоназальной дисплазии, так и синдрома Гольденхара [8, 9].
Рис. 1. Профиль плода с синдромом агнатии-голопрозэнцефалии в 12 нед беременности.
Рис. 2. Фенотип лица плода при синдроме агнатииголопрозэнцефалии в 12 нед беременности.
Гипоплазия нижней челюсти при различных нехромосомных синдромах обычно встречается в сочетании со скелетными дисплазиями и мышечно-скелетными аномалиями: синдром Пьера Робена (рис. 3), Тичера - Коллинза (Франческетти), акрофасциальный дизостоз, цереброкостомандибулярный синдром, ахондрогенез (рис. 4), ателостеогенез, кампомелическая дисплазия, диастрофическая дисплазия (рис. 5), синдром множественных птеригиумов, синдром Пены - Шокейра и др. Наличие микрогнатии характерно для синдрома Карпентера, синдрома Фринса, синдрома Меккеля - Грубера, гидролетального синдрома, синдрома Миллера - Дикера, синдрома Нунан, синдрома Секкеля, Рубинштейна - Тейби и др. Большинство из описанных синдромов имеют аутосомно-рецессивный либо аутосомно-доминантный тип наследования [10, 11].
Рис. 3. Микрогнатия у плода при синдроме Пьера Робена в 13 нед беременности.
Рис. 4. Микрогнатия у плода с ахондрогенезом в 13 нед беременности.
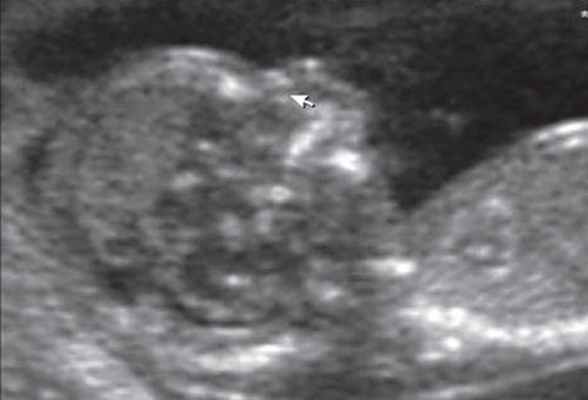
Рис. 5. Микрогнатия у плода с синромом диастрофической дисплазии в 12 нед беременности.
Пренатальная оценка положения и размера нижней челюсти может быть как субъективной, так и объективной. Так, на сегодняшний день известны оценки разных индексов измерения нижней челюсти [3, 6, 12], угла нижней челюсти [13, 14]. Эти измерения сопряжены со значительными погрешностями и в клинической практике применяются не часто, ввиду отсутствия стандартизации изучаемых срезов, трудоемкости и затратности обследования. Учитывая огромную значимость этого маркера, как для диагностики хромосомных, так и нехромосомных синдромов и ассоциаций, поиск новых объективных критериев микрогнатии продолжается 15.
Для качественной оценки особенностей строения нижней челюсти в I триместре беременности специалистами МГО МОНИИАГ совместно с профессором W. Sepulveda (Чили) был изучен и впервые описан новый ультразвуковой признак нижнечелюстной промежуток (mandibular "gap"), визуализируемый при первом скрининговом ("генетическом") ультразвуковом исследовании [18, 19].
Методика базируется на изучении коронарного скана лица плода, так называемого ретроназального треугольника, при котором визуализируется верхняя и нижняя челюсть. Техника получения этого скана чрезвычайно проста и может быть рекомендована для скринингового исследования в 11-14 нед беременности. Эта методика позволяет оценить нижнюю челюсть плода без применения трудоемких оценок, и не сопряжена с математически сложными расчетами коэффициентов, также она существенно не увеличивает время осмотра. Коронарный скан можно оценивать как в режиме 2D, так и в режиме объемной эховизуализации 3D. Методика оценки коронарного скана лица плода в I триместре беременности показана на рисунке 6.
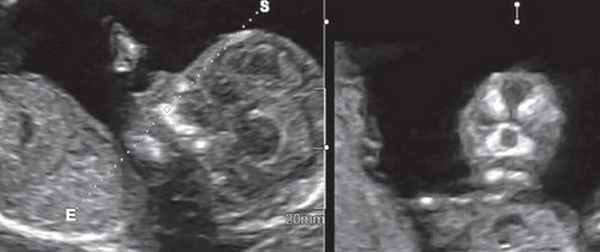
Рис. 6. Методика оценки коронарного скана лица плода в I триместре беременности.
Обе ветви нижней челюсти при сроке 11-14 нед беременности выглядят гиперэхогенными, а в месте слияния имеют характерный гипоэхогенный промежуток, ультразвуковой "разрыв". Этот признак визуализируется при нормальном развитии нижней челюсти (mandibular "gap").
Такие особенности ультразвуковой анатомии связаны с этапами эмбрионального развития костей нижней челюсти, ветви которой начинают развиваться из первой жаберной дуги с 7-й недели эмбрионального развития (рис. 7), и, постепенно приближаясь друг к другу к концу I триместра (на 14-й неделе беременности), образуют синостоз в области подбородка.
Размер этого промежутка уменьшается с увеличением срока беременности. Нижнечелюстной промежуток здорового плода представлен на рисунке 8.
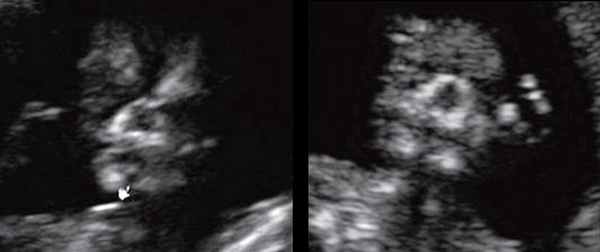
Рис. 8. Нижнечелюстной промежуток в 13 нед беременности при нормальном развитии нижней челюсти.
При патологии нижней челюсти (микрогнатии) в срок 11-14 нед беременности при изучении коронарного скана лица нижнечелюстной "промежуток" отсутствует, нижняя челюсть представлена единой, слившейся костной массой. Отсутствие нижнечелюстного "промежутка" (mandibular "gap") при эхографии в этот срок является маркером гипоплазии нижней челюсти (микрогнатии). Варианты отсутствия нижнечелюстного промежутка при микрогнатии при различных синдромах в срок 11-14 нед беременности представлены на рисунке 9.
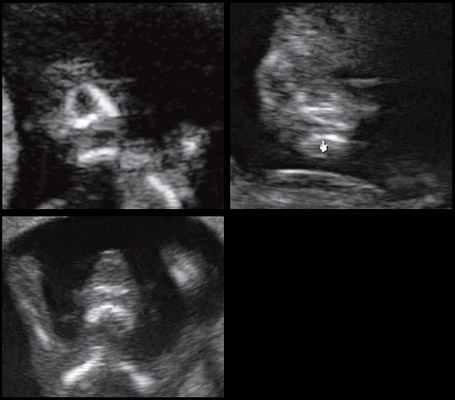
Рис. 9. Отсутствие нижнечелюстного промежутка при микрогнатии, при различных синдромах нехромосомного генеза в 11-14 нед беременности.
Оценка описанного признака при проведении пренатальной эхографии I триместра беременности не только проста в методологии и не требует больших затрат по времени исследования, но и высоко информативна, диагностически точна и специфична.
Литература
- Bianchi D.W., Crombleholme T.M., D'Alton M.E. Micrognathia. In Fetology: Diagnosis and Management of the Fetal Patient // McGraw-Hill: New York. 2000. P. 233-238.
- Nicolaides K.H., Salvesen D.R., Snijders R.J.M., Gosden C. Micrognathia fetal facial defects: Associated malformations and chromosomal abnormalities // Fetal Diagn Ther. 1993. V. 8. Р. 1-9.
- Paladini D., Morra T., Teodoro A., Lamberti A., Tremolaterra F., Martinelli P. Objective diagnosis of micrognathia in the fetus: the Jaw Index // Obstet Gynecol. 1999. V. 93. Р. 382-386.
- Dixon A.D., Hoyte D., Rоnning O. Prenatal development of the facial skeleton // In Fundamentals of Craniofacial Growth. CRC Press: Boca Raton. New York. 1997. Р. 59-97.
- Blaas H.G.K., Eriksson A.G., Salvesen K.A. et al. Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases // Ultrasound Obstet. Gynecol. 2002. V. 19. 1. P. 24-38.
- Paladini D. Fetal micrognathia: almost always anominous finding // Ultrasound Obstet. Gynecol. 2010. V. 35. P. 377-384.
- Cohen M.M.Jr. Perspectives on holoprosencephaly: Part I. Epidemiology, genetics, and syndromology // Teratology. 1989. V. 40. Р. 211-235.
- Carey J.C., Yong S.L. Frontonasal dysplasia and Goldenhar syndrome: the oculo-auriculo-frontonasal syndrome // Paper presented at the Conference on Malformations and Morphogenesis (March of Dimes). Dartmouth College, Hanover, NH, USA. 1981.
- Casey H.D., Braddock S.R., Haskins R.C., Carey J.C., Morales L. Frontonasal malformation and the oculoauriculovertebral spectrum: the oculoauriculofrontonasal syndrome // Cleft Palate Craniofac. J. 1996. V. 33. Р. 519-523.
- Ван Фехт Дж. Ультразвуковые маркеры хромосомных аномалий у плода // Ультразвуковая Диагностика. 1997. 3. С. 37-44.
- Turner G.M., Twining P. The facial profile in the diagnosis of fetal abnormalities // Clin Radiol. 1993. V. 47. Р. 389-395.
- Chitty L.S., Campbell S., Altman D.G. Measurements of the fetal mandible feasibility and construction of a centile chart // Prenat Diagn. 1993. V. 13. Р. 749-756.
- Otto C., Platt L.D. The fetal mandible measurement: an objective determination of fetal jaw size // Ultrasound Obstet Gynecol. 1991. V. 1. Р. 12-17.
- Rotten D., Levaillant J.M., Martinez H., Ducou H., Le Pointe D., Vicaut E. The fetal mandible: a 2D and 3D sonographic approach to the diagnosis of retrognathia and micrognathia // Ultrasound Obstet. Gynecol. 2002. V. 19. Р. 122-130.
- Bronshtein M., Blazer S., Zalel Y., Zimmer E.Z. Ultrasonographic diagnosis of glossoptosis in fetuses with Pierre Robin sequence in early and mid pregnancy // Am. J. Obstet. Gynecol. 2005. V. 193. Р. 1561-1564.
- Chitty L.S., Campbell S., Altman D.G. Measurements of the fetal mandible feasibility and construction of a centile chart // Prenat Diagn. 1993. V.13. Р. 749-756.
- Watson W. J., Katz V.L. Sonographic measurement of the fetal mandible: standards for normal pregnancy // Am J Perinatol. 1993. V. 10. Р. 226-228.
- Sepulveda W., Wong A., Andreeva E., Adzehova N. Absent mandibular gap at retronasal triangle view: a clue to the diagnosis of micrognathia in the first trimester // Ultrasound in obstetrics and gynecology. 2012. V. 39. P. 152-156.
- Sepulveda W., Wong A., Andreeva E. et al. A novel, simple technique for diagnosis of micrognathia in firsttrimester: identification of the receding chin on the retronasal triangle (RNT) view. Oral poster abstracts. 21 World Congress on Obstetrics and Gynecology. LosAngeles // Ultrasound in Obstetrics and Gynecology. 2011. V. 38. P. 64.
УЗИ сканер HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Врожденные патологии конечностей
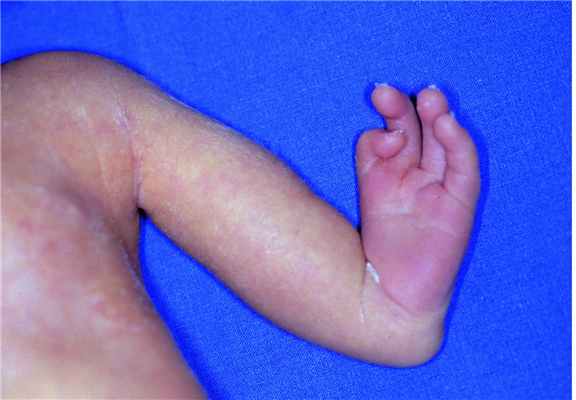
Врожденные пороки конечностей включают отсутствие или неполное развитие конечностей, лишние конечности или ненормально развитые конечности, что присутствуют с рождения.
Неполноценности конечностей
Врожденные ампутации и недостатки конечностей – это отсутствие или неполноценность конечностей при рождении. Общая распространенность составляет 7,9/10000 живорожденных детей. Большинство из них возникает вследствие первичного внутриутробного торможения роста или как нарушение, вторичное к внутриутробному разрушению нормальных эмбриональных тканей. Чаще всего страдают верхние конечности.
Дефициты конечностей могут быть:
Продольные дефициты включают специфичные нарушения развития (например, полное или частичное отсутствие лучевой, малоберцовой или большеберцовой костей). Отсутствие лучевой кости является наиболее распространенным недостатком верхних конечностей, а гипоплазия малоберцовой – наиболее распространенным недостатком нижних конечностей. Около двух третей случаев связаны с другими врожденными нарушениями, в том числе, с синдромом Адамса–Оливера (врождённое недоразвитие кожи с частичной аплазией костей черепа и терминальными поперечными мальформациями конечностей), синдромом Холта–Орама, TAR-синдромом (тромбоцитопения и аплазиялучевой кости), анемией Фанкони и VACTERL-синдромом (позвоночные аномалии, анальная атрезия, пороки развития сердца, трахеоэзофагеальный свищ, почечные аномалии, радиальная аплазия и аномалии конечностей).
При поперечных недостатках,, все элементы конечности выше определенного уровня отсутствуют, а сами конечности напоминают ампутационную культю. Амниотические перетяжки являются наиболее распространенной причиной; степень дефицита варьируется в зависимости от локализации перетяжки, и, как правило, нет никаких других дефектов или аномалий. Остальные случаи, в основном, возникают из-за скрытых генетических синдромов, таких как синдром Адамса-Оливера, или хромосомных аномалий.
При поперечном или продольном дефиците, в зависимости от этиологии, младенцы могут также иметь гипопластические или раздвоенные кости, синостозы, дупликации, вывихи или другие дефекты костей; например, при проксимальном очаговом дефиците бедренной кости проксимальный отдел бедренной кости и вертлужная впадина не развиваются. Могут быть затронуты одна или более конечностей, и тип дефекта может быть разным в каждой конечности. Аномалии центральной нервной системы наблюдаются редко.
Полидактилия
Полидактилия – это наличие дополнительных пальцев; она является наиболее частой врожденной деформацией конечностей. Эта деформация классифицируется на преаксиальную, центральную и постаксиальную.
Преаксиальная полидактилия – это дополнительный большой палец на руке или ноге. Проявления варьируют от широкой или дублированной дистальной фаланги до полного дублирования пальца. Это может наблюдаться как отдельное проявление, возможно, с аутосомно-доминантным наследованием, или может быть частью некоторых генетических синдромов, в том числе акрокаллозального синдрома (с задержкой развития и дефектами мозолистого тела), синдромов Карпентера и Пфайффера (с краниосиностозом Краниосиностоз Краниосиностоз является преждевременным слиянием одного или более черепных швов. (См.также Введение в врожденные черепно-лицевые и скелетно-мышечные расстройства (Introduction to Congenital. Прочитайте дополнительные сведения ).
Центральная полидактилия- редкое явление, что включает в себя дублирование второго, третьего и четвертого пальцев. Это может быть связано с синдактилией и расщеплённой кистью. Большинство случаев синдромные.
Постаксиальная полидактилия является наиболее распространенной и включает в себя дополнительный палец на локтевой/малоберцовой стороне конечности. Чаще всего, дополнительный палец является рудиментарным, но может быть полностью развит. У лиц африканского происхождения этот тип полидактилии, как правило, возникает как единичный дефект. В других популяциях он чаще связан с синдромом множественных врожденных аномалий или хромосомными дефектами. Возможные синдромы включают синдром цефалополисиндактилии Грейга, синдром Меккеля, синдром Эллиса-Ван Кревельда, синдром МакКузика-Кауфмана, синдром Дауна Синдром Дауна (трисомия 21) Синдром Дауна является аномалией 21-й хромосомы, может проявляться нарушением умственного развития, микроцефалией, небольшим ростом и характерным внешним видом. Диагноз предполагают на основании. Прочитайте дополнительные сведенияСиндактилия
Синдактилия – это перепончатость или сращивание пальцев рук или ног. Определены несколько различных типов, и большинство передаётся по аутосомно-доминантному типу наследования. Простая синдактилия включает в себя только сращивание мягких тканей, в то время как сложная синдактилия также включает сращивание костей. Сложная форма синдактилии присутствует при синдроме Аперта (с краниосиностозом Краниосиностоз Краниосиностоз является преждевременным слиянием одного или более черепных швов. (См.также Введение в врожденные черепно-лицевые и скелетно-мышечные расстройства (Introduction to Congenital. Прочитайте дополнительные сведенияДиагностика врожденных аномалий конечностей
Обычно используется рентгенография
Иногда генетические тесты
Обычно проводится рентгенография для определения того, какие кости затронуты патологией. Если дефект является семейным или есть подозрения на генетический синдром, оценка должна также включать в себя тщательную проверку на другие физические, хромосомные и генетические аномалии. По возможности, стоит провести консультацию у клинического генетика.
Лечение врожденных аномалий конечностей
Лечение полидактилии и синдактили производится хирургическим путем. Лечение отсутствия или гипоплазии конечностей заключается, в основном, в протезировании, которое является наиболее целесообразным при отсутствии нижних конечностей и полном или почти полном отсутствии верхних конечностей. Если в руке или кисти сохраняется какая-либо активность, независимо от тяжести нарушения развития, стоит провести тщательную оценку функциональности до рекомендации протезирования или хирургического вмешательства. Терапевтическую ампутацию конечности или любого участка конечности следует рассматривать только после оценки функциональных и психологических последствий ее потери, и в тех случаях, когда ампутация необходима для установки протеза.
Протезы верхних конечностей должны быть разработаны так, чтобы они могли выполнять как можно больше функций и чтобы число используемых устройств было сведено к минимуму. Дети используют протез наиболее успешно, когда он установлен рано и становится неотъемлемой частью их тела и внешнего вида при взрослении. Приспособления, используемые в младенчестве, должны быть максимально простыми и долговечными; например, крюк предпочтительнее, чем биоэлектрическая рука. При эффективной ортопедической и вспомогательной поддержке большинство детей с врожденными ампутациями ведут нормальный образ жизни.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: