Синдром Лежена. Синдром крик кошки в неврологии
Добавил пользователь Владимир З. Обновлено: 31.01.2026
Синдром Лежена. Синдром крик кошки в неврологии
Врожденный комплекс пороков развития, обусловленный нехваткой генов, локализованных в коротком плече 5-й хромосомы, описан в 1963 г. J. Lejeune и др. Частота синдрома среди новорожденных не установлена, одинаково часто поражаются мальчики и девочки.
В большинстве случаев нехватка (5р—) возникает вследствие простой делеции хромосомы в гаметах одного из родителей. Реже она обусловлена сбалансированной транслокацией с участием короткого плеча 5-й хромосомы, носителем которого является отец или мать.
Размеры недостающего участка хромосомы в клетках больного могут быть различны, с чем, по-видимому, связана некоторая вариабельность клинических проявлений синдрома. Обычно дети рождаются с низкой массой (до 2500 г) даже при доношенной беременности. Наиболее постоянным симптомом, от которого синдром получил свое название, является специфический тембр голоса, напоминающий кошачье мяуканье. Симптом обусловлен особенностями строения гортани, определяемыми ларингоскопически — маленьким вялым надгортанником, имеющим тенденцию опускаться над голосовой щелью, при этом сами голосовые связки не изменены. Описанная особенность строения гортани проявляется рентгенологически уменьшением воздушного пространства над голосовыми связками. С возрастом особенность крика исчезает, но часто остается стридор и склонность к инфекционным заболеваниям верхних дыхательных путей, а также такие симптомы, как круглое луноподобное лицо, косой разрез глаз с опущенными наружными углами, эпикант, гипертелоризм, несколько уплощенный нос, низко расположенные ушные раковины, впереди которых имеются небольшие (размером 1 : 3 мм) круглые фиброзные узелки. Мозговой череп относительно малых размеров (микроцефалия), долихоцефальной формы, с выступающими лобными буграми. Обычно внешние особенности дополняются маленькой ретрагированной нижней челюстью и короткой шеей с избыточной кожей, формирующей крыловидные складки. В некоторых случаях может быть расщепление губы и (или) твердого нёба либо высокое готическое нёбо и расщепление мягкого нёба. Офтальмологические нарушения проявляются в виде альтернирующего или постоянного косоглазия, астигматизма. Среди аномалий развития внутренних органов наиболее часты пороки развития сердца и сосудов, почек. У мальчиков часто бывает гипоспадия. Общая мышечная гипотония, характерная для новорожденных с синдромом Лежена, обычно сохраняется в течение года и более. Больные отстают в психомоторном и физическом развитии.

Биохимические нарушения при синдроме Лежена неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная аминоацидемия и аминоацидурия.
Дерматоглифически в большинстве случаев определяется поперечная борозда на обеих ладонях, дистальный трирадиус отсутствует или его ось находится в положении t.
Средняя продолжительность жизни больных снижена. Больные погибают вследствие сердечной или почечной недостаточности, либо от различных интеркурентных инфекционных заболеваний.
При цитогенетическом исследовании обычно выявляется делеция короткого плеча 5-й хромосомы. Иногда 5-я хромосома при рутинном исследовании может казаться интактной или ее короткие плечи могут быть даже увеличенными. С помощью методов дифференциальной окраски хромосом в этих случаях обычно удается установить, что в действительности часть короткого плеча 5-й хромосомы представлена фрагментом другой хромосомы, локализованным в необычном месте в результате транслокации. Может быть кольцевая хромосома 5.
Заболевание следует дифференцировать от других врожденных аномалий развития хромосомной и нехромосомной этиологии. Нозологический диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом группы В (авторадиографии, флюоресцирующих красителей или специфической обработки для получения «бэндинга»).
Синдром кошачьего глаза ( Синдром Шмида-Фраккаро )
Синдром кошачьего глаза — это редкое генетическое заболевание, которое характеризуется нарушениями развития глаз, лицевого скелета, разнообразными аномалиями внутренних органов. Патология возникает, когда в кариотипе человека есть так называемая дополнительная маркерная хромосома. Синдром проявляется патогномоничной триадой признаков: колобомой глазной радужки, атрезией ануса, деформациями ушных раковин. Часто наблюдаются пороки формирования сердца, почек, печени, у части больных также встречается умственная отсталость. Диагностика синдрома основана на цитогенетическом исследовании. Лечение симптоматическое: кардиохирургическая помощь, пластическая коррекция дефектов лица.
МКБ-10
Общие сведения
Синдром кошачьего глаза (в иноязычной литературе ‒ «cat eye syndrome», CES) имеет синонимичное название синдром Шмида-Фраккаро. Его клинические признаки впервые описаны шведским офтальмологом Отто Хаабом в 1898 г., а генетические основы установлены только в 1965 г. Патология встречается крайне редко: 1 случай на 50-150 тыс. живорожденных новорожденных. Однако проблема не теряет своей актуальности в современной генетике, поскольку от специалистов требуется максимально раннее выявление болезни, подбор адекватного комплексного лечения.
Причины
Развитие синдрома кошачьего глаза связывают с наличием в кариотипе пациента дополнительной маркерной хромосомы, которая образована из генетического материала 22 хромосомы путем инвертированной дупликации участка 22pter → q11. В результате возникает частичная тетрасомия по 22-й хромосоме. Также не исключены мозаичные формы синдрома. Мутация происходит спонтанно в период раннего эмбриогенеза. Конкретные провоцирующие факторы пока не установлены.
Патогенез
Хромосомная аномалия ассоциирована с нарушениями онтогенеза в эмбриональном периоде, когда происходит закладка всех органов и тканей. Формирование колобомы, вероятнее всего, обусловлено неправильным закрытием щели глазного бокала, вследствие чего нарушается форма радужной оболочки глаза. Формирование пороков сердца происходит на 3-8 неделях внутриутробного развития под влиянием генетических мутаций.
Симптомы
Типичные проявления синдрома кошачьего глаза определяются сразу после рождения. Младенец имеет специфические аномалии лица: брахицефальную форму головы, выступающий лоб, недоразвитие челюстей, сопровождающееся затруднениями в открывании рта. Также характерны расщелины мягкого и твердого неба, опущение век, асимметрия черт лица. У большинства больных наблюдается деформация ушной раковины, появление периаурикулярных отростков.
Патогномоничным признаком синдрома является двусторонняя колобома — дефект радужки, который придает сходство с кошачьим глазом. Также возникает атрезия ануса. Из соматических пороков распространены аномалии сердца: атрезия легочной артерии, тетрада Фалло, дефекты межпредсердной или межжелудочковой перегородки. Возможны поражения ЖКТ (свищи, дивертикулы, мегаколон), урогенитального тракта (гипоплазия почек, гидронефроз).
Тяжелые хромосомные нарушения при болезни кошачьего глаза ассоциированы с задержкой физического развития ребенка. Такие пациенты обычно имеют невысокий рост, что вызвано недостаточной выработкой соматотропина. Распространены нарушения темпов полового созревания у больных обоего пола, возможно формирование первичного гипогонадизма. В раннем детском возрасте беспокоят частые инфекции, связанные с недостаточностью иммунитета.
Осложнения
Около 7% эмбрионов с грубыми пороками погибают в первые месяцы внутриутробной жизни. При этом у матери происходит самопроизвольный аборт. Синдром кошачьего глаза, который сочетается со множественными аномалиями, имеет высокий показатель летальности и в постнатальном периоде. Основными причинами смерти являются кардиологические нарушения, присоединившиеся бактериальные инфекции.
Дефекты внешности, которые вызваны колобомой и аномалиями строения лица, зачастую затрудняют социализацию ребенка в коллективе, делают его объектом насмешек сверстников. На фоне этого могут возникать депрессии, аффективные состояния, психопатоподобные расстройства. Описаны случаи чрезмерной агрессии пациентов, направленной на окружающих. Около 32% больных страдает умственной отсталостью разной степени выраженности.
Диагностика
Обследованием ребенка занимается педиатр-неонатолог, по показаниям к осмотру привлекают невролога, кардиолога, генетика. Заподозрить наличие синдрома кошачьего глаза удается по патогномоничной триаде: атрезии анального отверстия, колобоме радужки, деформации ушей, которые есть у 41% больных. Расширенная диагностика включает лабораторно-инструментальные методики:
- Цитогенетическое исследование. Для подтверждения диагноза необходима флуоресцентная гибридизация с ДНК-зондом, с помощью которой обнаруживается характерная хромосомная мутация. Для исследования используется 72-часовая культура лимфоцитов периферической крови.
- Эхокардиография.УЗИ сердца с допплерографией — самый безопасный метод оценки структурно-функциональных особенностей органа и визуализации врожденных пороков. Для уточнения диагноза рекомендованы ангиография, КТ или МРТ сердца.
- КТ лицевого черепа. Исследование проводится для детальной визуализации аномалий костных структур, определения их глубины и локализации. При планировании ортодонтической помощи методика дополняется телерентгенографией.
- Дополнительные методы. Для диагностики нарушений глазного аппарата выполняется офтальмоскопия. При подозрении на пороки ЖКТ назначается обзорная рентгенография или КТ органов брюшной полости. Для выявления проблем с мочевыделительной системой применяется УЗИ почек, экскреторная урография.
Лечение синдрома кошачьего глаза
Специфическая терапия синдрома кошачьего глаза отсутствует. Больным необходима помощь разных специалистов в соответствии с характером и степенью тяжести клинических проявлений. Лечение направлено на предупреждение осложнений, улучшение качества жизни пациента, восстановление нормального фенотипа. Выделяют следующие направления медицинской помощи:
- Правильный уход. Младенцам необходима коррекция кормления, подбор ортодонтических сосок, поскольку грудное вскармливание затруднено из-за невозможности полного открывания рта.
- Кардиохирургическая коррекция. При сердечных пороках, сопровождающихся нарушениями кровообращения, рекомендовано хирургическое вмешательство на первом-втором году жизни ребенка, а критические нарушения оперируются в возрасте 3-6 месяцев.
- Пластические операции. Для закрытия дефектов неба, устранения асимметрии лица и других неэстетичных особенностей внешности требуется помощь пластических и челюстно-лицевых хирургов. Сроки операций устанавливаются индивидуально.
- Реабилитация. Для правильного развития речи с учетом аномалий строения артикуляционного аппарата показаны занятия с логопедом. В некоторых случаях назначается ортодонтическая коррекция.
Прогноз и профилактика
Пациенты с мягкими фенотипическими проявлениями синдрома кошачьего крика и отсутствием тяжелых соматических аномалий имеют нормальную продолжительность жизни. Менее благоприятный прогноз при кардиологических пороках, которым сопутствуют сердечная недостаточность, задержка физического развития. Профилактика синдрома включает медико-генетическое консультирование, пренатальную диагностику для пар с отягощенной наследственностью.
1. Синдром «кошачьих глаз» (психиатрический аспект)/ И.А. Макаров, С.Б. Гаврилина, Б.Г. Белозеров// Журнал неврологии и психиатрии им. С.С. Корсакова. — 2019. — №11.
2. Синдром «кошачьего глаза» без атрезии ануса, с амплификацией генов ada2 и il17ra (клинический случай)/ М.И. Душар, Г.Р. Акопян, Г.В. Макух, М.В. Влох// Современная педиатрия. — 2019. — №2.
3. Синдром кошачьего глаза у ребенка с врожденным пороком развития сердца (клинический случай)/ А.А. Музычина, И.А. Бугоркова, Е.О. Кальней// Медико-социальные проблемы семьи. — 2018. — №2.
4. Мозаичный вариант частичной трисомии по длинному плечу хромосомы 22 у ребенка с врожденным пороком сердца/ А.Г. Шаповалов, О.Б. Полодиенко, И.В. Иванова// Детский врач. — 2015. — №2.
Синдром кошачьего крика
Синдром «кошачьего крика» – хромосомное нарушение, обусловленное делецией (отсутствием) фрагмента короткого плеча 5-ой хромосомы. Плач новорожденных с синдромом «кошачьего крика» по звуку напоминает кошачье мяуканье, что и послужило названию патологии. Кроме этого, у детей имеет место микроцефалия, лунообразное лицо, косоглазие, аномалии прикуса, различные врожденные пороки, грубое интеллектуальное недоразвитие и т. д. Синдром «кошачьего крика» диагностируется на основании совокупности характерных признаков и цитогенетического исследования. Специфического лечения синдрома «кошачьего крика» не существует; дети могут нуждаться в хирургической коррекции тяжелых врожденных аномалий.
Синдром «кошачьего крика» (синдром Лежена) – частичная моносомия, связанная с нарушением структуры короткого плеча 5-ой хромосомы (потерей от 1/3 до 1/2 его длины, реже - полной утратой короткого плеча). Синдром «кошачьего крика» относится к числу редких хромосомных заболеваний с популяционной частотой 1:45-50 тыс. Среди новорожденных с синдромом «кошачьего крика» отмечается преобладание девочек над мальчиками в соотношении 4:3. Заболевание было описано в 1963 г. французским генетиком и педиатром Ж. Леженом и по автору получило название «синдром Лежена». Однако в литературе за данной патологией закрепилось образное название, связанное со специфическим признаком – плачем новорожденных, напоминающим кошачий крик.
Причины синдрома «кошачьего крика»
Развитие синдрома «кошачьего крика» связано с потерей фрагмента 5-ой хромосомы, а, следовательно, генетической информации, хранящейся на этом участке. В 85-90% случаев делеция короткого плеча образуется в результате случайной мутации, в 10-15% наследуется от родителей, являющихся носителями сбалансированной транслокации.
Наиболее частыми цитогенетическими вариантами хромосомной аберрации служат утрата одной трети или половины длины короткого плеча 5-ой хромосомы. Потеря меньшего участка или всего плеча встречается исключительно редко. При этом для степени выраженности клинической картины синдрома «кошачьего крика» важен не размер утерянного фрагмента, а отсутствие конкретного участка хромосомы. Так, при потере небольшого участка хромосомы в области 5p15.2 развиваются все клинические признаки синдрома, кроме кошачьего крика; критическим для возникновения характерного крика является выпадение участка хромосомы в области 5p15.3.
Наряду с простой делецией, могут встречаться другие цитогенетические вариации синдрома «кошачьего крика»: мозаицизм, кольцевая 5-я хромосома с делецией участка короткого плеча, реципрокная транслокация короткого плеча 5-ой хромосомы на другую хромосому.
Непосредственной причиной мутации могут выступать различные повреждающие факторы, воздействующие на половые клетки родителей либо на зиготу (алкоголь, курение, наркотические вещества, ионизирующая радиация, лекарственные препараты, химикаты и пр.). Вероятность появления ребенка с синдромом «кошачьего крика» выше в семьях, где уже рождались дети с подобным заболеванием.
Симптомы синдрома «кошачьего крика»
Новорожденные с синдромом «кошачьего крика», как правило, рождаются доношенными, но с небольшой пренатальной гипотрофией (средняя масса при рождении около 2500 г). Беременность у матери может протекать абсолютно нормально или сопровождаться угрозой самопроизвольного прерывания не чаще, чем в популяции. Наиболее патогномоничным ранним признаком синдрома является плач ребенка, который напоминает мяуканье кошки. Высокое и пронзительное звучание детского крика обусловлено анатомическими особенностями строения гортани при данном синдроме – узостью ее просвета, небольшим надгортанником, необычной складчатостью слизистой оболочки, мягкой консистенцией хрящей. Некоторые авторы считают, что специфический крик имеет центральное происхождение и не связан с недоразвитием гортани. Примерно у трети детей «кошачий крик» исчезает к 2-м годам, у остальных остается на всю жизнь.
Фенотип детей с синдромом «кошачьего крика» отличается преобладанием лицевой части черепа над мозговой, лунообразным лицом, гипертелоризмом, антимонголоидным разрезом глаз, эпикантом, деформацией ушных раковин, плоской спинкой носа, короткой шеей с крыловидными складками. При обследовании у детей выявляется микроцефалия, мышечная гипотония, снижение рефлексов, нарушение сосания и глотания. В неонатальном периоде может развиваться инспираторный стридор и цианоз.
Другие клинические проявления синдрома «кошачьего крика» могут значительно варьироваться по своему сочетанию у отдельных больных. Со стороны зрительной системы нередко обнаруживается врожденная катаракта, близорукость, косоглазие, атрофия зрительного нерва. Изменения со стороны костно-мышечной системы проявляются синдактилией стоп, врожденным вывихом бедра, косолапостью, плоскостопием, клинодактилией V пальца кисти, сколиозом, диастазом прямых мышц живота, паховыми и пупочными грыжами. Частыми спутниками синдрома «кошачьего крика» являются нарушения прикуса, «готическое» нёбо, микрогения, расщелины нёба и верхней губы, расщепление язычка.
У многих пациентов наблюдаются врожденные пороки сердца (ДМЖП, ДМПП, открытый артериальный поток, тетрада Фалло), пороки развития почек (гидронефроз, подковообразная почка), крипторхизм, гипоспадия. Реже отмечается мегаколон, запоры, кишечная непроходимость. Дерматоглифическими признаками синдрома «кошачьего крика» могут служить одна ладонная складка, поперечные складки сгибания и др.
Поведение детей характеризуется гиперактивностью, однообразными движениями, склонностью к агрессии и истерикам. Детям с синдромом «кошачьего крика» свойственна глубокая умственная отсталость в степени имбецильности и идиотии, грубое системное недоразвитие речи, выраженное отставание в моторном и физическом развитии.
Половая и репродуктивная функции у лиц с синдромом «кошачьего крика» обычно не страдают. Иногда у женщин выявляется двурогая матка, у мужчин – уменьшение размеров тестикул, однако сперматогенез существенно не нарушен, как, например, при синдроме Клайнфельтера.
Диагностика синдрома «кошачьего крика»
Если в семье уже имелись случаи хромосомных заболеваний, еще на этапе планирования беременности будущим родителям рекомендуется посетить генетика и пройти генетическое тестирование. В процессе беременности наличие у плода синдрома «кошачьего крика» может быть заподозрено на основании результатов ультразвукового пренатального скрининга. В этом случае для окончательного подтверждения хромосомной аномалии рекомендуется проведение инвазивной пренатальной диагностики (амниоцентеза, биопсии ворсин хориона или кордоцентеза) и непосредственного анализа генетического материала плода.
После рождения предварительный диагноз синдрома «кошачьего крика» устанавливается неонатологом на основании типичных диагностических признаков (характерного плача, фенотипических черт, множественных стигм дизэмбриогенеза). Для подтверждения хромосомной патологии проводится цитогенетическое исследование.
Учитывая наличие у детей с синдромом «кошачьего крика» множественных аномалий развития, необходимо, чтобы в первые дни жизни новорожденные были осмотрены детским кардиологом, детским офтальмологом, детским урологом, детским ортопедом и другими специалистами.
Лечение синдрома «кошачьего крика»
Специфического лечения данного хромосомного заболевания в настоящее время не существует. Для стимуляции психомоторного развития под наблюдением детского невролога проводятся курсы медикаментозной терапии, массажа, физиотерапии, ЛФК. Дети с синдромом «кошачьего крика» нуждаются в помощи психологов, дефектологов, логопедов.
Врожденные пороки сердца при синдроме «кошачьего крика» часто требуют хирургической коррекции, поэтому детям необходима консультация кардиохирурга, проведение ЭхоКГ и других необходимых исследований. Дети с патологией мочевыделительной системы должны находиться под наблюдением детского нефролога и периодически проходить комплекс необходимых обследований (УЗИ почек, общий анализ мочи, биохимическое исследование крови и мочи и др.).
Прогноз и профилактика синдрома «кошачьего крика»
На продолжительность и качество жизни больных влияет тяжесть самого синдрома и сопутствующих врожденных пороков, уровень оказания медицинской и психолого-педагогической помощи. В целом долговременный прогноз неблагоприятный. При специальном обучении дети имеют словарный запас, достаточный для бытового общения, однако по уровню психомоторного развития обычно не поднимаются выше дошкольников.
Профилактика синдрома «кошачьего крика» заключается в тщательной подготовке к беременности и исключении возможных неблагоприятных воздействий на организм родителей еще задолго до зачатия. При рождении в семье ребенка с синдромом «кошачьего крика», родители в обязательном порядке должны пройти цитогенетическое обследование для исключения носительства реципрокной сбалансированной транслокации.
Синдром Айкарди
Синдром Айкарди — это Х-сцепленное генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела с формированием хориоретинальных лакун и вариабельными эмбриональными аномалиями скелета. Типичной клинической картиной выступает триада признаков: инфантильные спазмы, задержка психического развития, нарушения зрения. Диагностика осуществляется при помощи электроэнцефалографии, церебральной МРТ, офтальмоскопии, изучения зрительных потенциалов, рентгенографии. Генетические методы диагностики пока не найдены. Лечение паллиативное с назначением комбинаций антиконвульсантов для постоянного приема, периодических курсов кортикостероидов.
Синдром Айкарди относится к редким расстройствам эмбрионального развития нервной системы. Впервые описан в 1965 году французским детским неврологом Жаном Айкарди как заболевание, характеризующееся следующей триадой признаков: агенезия мозолистого тела, офтальмологические расстройства, мышечные спазмы. Название синдрому в честь Ж. Айкарди было дано в 1972 г. Дальнейшее его изучение расширило список характерных клинических признаков, включив в них лицевой дисморфизм, умственную отсталость, аномалии позвоночного столба. Развитие методов нейровизуализации и генетических исследований позволило сделать диагностику более точной. По данным мировой статистики, всего насчитывается около 500 заболевших. Синдром связан с патологией Х-хромосомы, встречается исключительно у девочек. У плодов мужского пола возникают аномалии, не совместимые с жизнью.
Заболевание является генетической патологией, связанной с нарушением участка на Х-хромосоме. Считается, что оно имеет доминантное сцепленное с Х-хромосомой наследование. Теоретически вероятность рождения больной девочки от матери, страдающей синдромом, составляет 50%. Наличие патологической Х-хромосомы у мальчиков приводит к внутриутробной смерти.
На практике генетическая передача не зафиксирована, поскольку заболевшие девочки не могут иметь потомства в силу выраженных психомоторных расстройств. Все исследованные случаи синдрома Айкарди возникали de novo. Встречаются преимущественно спорадические варианты заболевания. Исключение составляет единственный известный случай у двух сестер. В связи с малым количеством заболевших, проведение достоверного исследования мутагенных триггеров затруднительно.
Патогенетические механизмы развития заболевания находятся в стадии изучения. Предполагается, что мутации генов обуславливают расстройство нейрональной миграции, происходящей в период между 12-й и 24-й неделями эмбрионального развития. В эмбриогенезе миграция неокортикальных нейронов на периферию приводит к образованию коры и подкорковых зон. Ее нарушения обуславливают дисгенезию мозолистого тела, аномальное формирование извилин и архитектоники мозга.
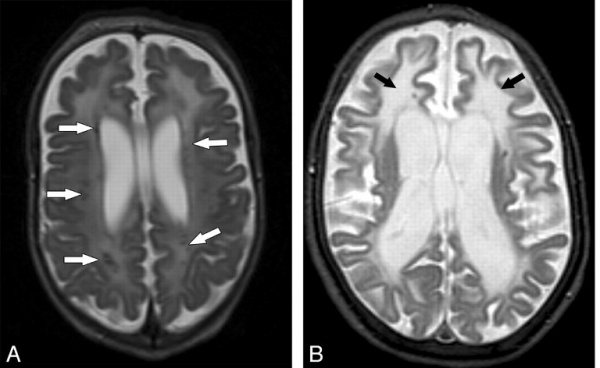
В норме в мозолистом теле проходят межполушарные проводящие пути, поэтому его дисэмбриогенез влечет нарушение связей между полушариями. Макроскопически в головном мозге выявляется микрогирия, субатрофия церебральной коры, перивентрикулярные кисты, гетеротопия, агенезия мозолистого тела. Микроскопически в сетчатке отмечается истончение слоев, уменьшение количества сосудов, гиперплазия пигментного эпителия.
Дети с синдромом Айкарди рождаются без видимых патологий, обычно в срок. В большинстве случаев беременность и роды протекают без осложнений. Заболевание манифестирует инфантильными спазмами в первые месяцы жизни. Спазмы представляют собой фокальные эпиприступы, при патологии Айкарди отличаются большой вариабельностью.
Возможны миоклонии, тонические судороги, ретропульсии, пропульсии. Наблюдаются симметричные и асимметричные, единичные и серийные судорожные приступы, типична тенденция к серийности. Флексорные спазмы отмечаются у 97% пациенток. В 42% случаях заболевания инфантильные спазмы сочетаются с другими видами эпиприступов, как фокальными, так и генерализованными. Фокальные припадки происходят с вовлечением преимущественно лицевой мускулатуры.
Задержка психомоторного развития характерна для всех больных синдромом. Отмечается обедненность эмоциональных реакций, утрачен контакт с окружающими, останавливается развитие навыков и предречевое развитие. Позже присоединяются моторные расстройства: геми- и тетрапарезы с повышением или снижением тонуса мышц, возможна спастичность. В отдельных случаях описана умеренная задержка развития, легкая степень олигофрении.
Офтальмологические аномалии вариабельны, включают: атрофию зрительного нерва, микроофтальмию, колобому. Возможна катаракта, пигментный ретинит. Клинически наблюдается значительное снижение зрительной функции. Со стороны желудочно-кишечного тракта характерна неустойчивость стула, проблемы с кормлением, гастроэзофагальный рефлюкс.
Среди челюстно-лицевых дисморфий описаны асимметрия лица, гипертелоризм, аномалии ушной раковины, выступающие резцы. У 33% пациенток отмечаются дисэмбриогенетические изменения позвоночника и ребер в виде полупозвонков, расщепления позвоночника, агенезии ребра, аномального реберно-позвоночного сочленения. В возрасте после 7 лет типично замедление роста, в пубертате — задержка полового развития.
Гемангиомы, невусы и другие дерматологические проявления описаны у 20% больных. В 7% случаев синдром Айкарди протекает с аномалиями конечностей: гипоплазией пальцев, синдактилией, каптодактилией.
Выраженная олигофрения, двигательные расстройства с первых месяцев жизни обуславливают глубокую инвалидность ребенка. Девочки нуждаются в постоянном уходе, половина из них неспособны к элементарному самообслуживанию. Нарушение мышечной иннервации при отсутствии постоянных реабилитационных мероприятий сопровождается гипотрофиями, в случаях спастичности — развитием контрактур.
Судорожный синдром с генерализованными припадками и кластерными приступами усугубляет неврологический дефицит, олигофрению. Позвоночные аномалии приводят к развитию выраженного сколиоза. Больные с синдромом Айкарди подвержены рецидивирующей пневмонии, имеют больший риск возникновения новообразований.
Внешний вид ребенка с лицевыми аномалиями, уменьшенным кончиком носа, латерально расположенными бровями позволяет врачу предположить генетическое заболевание. В неврологическом статусе показательны мышечная гипотония или односторонняя спастичность, снижение мышечной силы в конечностях, оживление рефлексов, снижение психического развития. Диагностика синдрома осуществляется совместно с генетиками, однако специфический генетический анализ еще не разработан. Назначаются следующие исследования:
- Электроэнцефалография. Наиболее типичным ЭЭГ-признаком синдрома Айкарди выступает гипсаритмия, появляющаяся после манифестиции инфантильных спазмов. Типично различие ЭЭГ-паттерна двух полушарий, феномен «расщепленного мозга». У ряда пациенток энцефалографическая картина имеет стертый характер. Наличие вариабельных судорожных припадков обуславливает полиморфизм изменений ЭЭГ.
- Офтальмоскопия. Выявляет наличие хориоретинальных лакун, визуализирующихся как белесоватые депегментированные участки сетчатки. Двусторонние лакуны расположены асимметрично, в 10-20% случаев очаги диагностируются только в одном глазу. При офтальмоскопии пациенток более старшего возраста диагностируется катаракта, колобома и/или атрофия диска зрительного нерва.
- Зрительные вызванные потенциалы (ЗВП). Исследуются для определения остроты зрения в младенческом возрасте и проведения дифдиагностики с другими офтальмологическими заболеваниями. У маленьких пациенток без изменений на глазном дне острота зрения может соответствовать возрастной норме.
- МРТ головного мозга. Визуализирует полное или частичное недоразвитие мозолистого тела, признаки корковой атрофии, пахигирию, расширение борозд. III желудочек расширен, вместе с боковыми желудочками смещен вверх, вокруг него выявляются церебральные кисты. Отмечается изменение формы латеральных желудочков, множественные области гетеротопии с отсутствием дифференцировки слоев коры, гидроцефалия.
- Рентгенография скелета. Необходима для диагностики костных аномалий, сопровождающих синдром Айкарди.
Перинатальная ДНК-диагностика не разработана. Выявление патологии возможно в ходе ультразвукового исследования беременной. По данным УЗИ 1 триместра возможна диагностика агенезии мозолистого тела. Позже выявляются микроцефалия, интракраниальные кисти, позвоночные аномалии, прочие пороки развития.
Дифференциальная диагностика
Необходимо дифференцировать патологию Айкарди от прочих ранних мультисистемных поражений, включающих сочетание дисгенеза церебральных структур и нарушения строения сетчатки. Первым симптомом септооптической дисплазии выступает горизонтальный нистагм, отсутствующий у пациентов с синдромом Айкарди. Отличительной чертой туберозного склероза служит наличие дерматологических симптомов: гиперпигментаций, ангиофибром, участков «шагреневой» кожи. Папилло-ренальный синдром протекает с поражением почек. Дифференциальная диагностика с врожденными TORH-инфекциями проводится на основании данных лабораторных обследований.
Следует отличать синдром Айкарди от заболевания Айкарди-Гутиера с аутосомно-рецессивным типом наследования. Последнее относится к прогрессирующим лейкодистрофиям детского возраста, характеризуется потерей приобретенных навыков, лимфоцитозом цереброспинальной жидкости, повышенным уровнем альфа-интерферона, кальцификатами в подкорковых структурах.
Лечение синдрома Айкарди
В основе терапии заболевания лежит паллиативная медикаментозная помощь и реабилитационные мероприятия. Базовой схемой является сочетание противосудорожных и гормональных фармпрепаратов, что позволяет добиться снижения частоты эпиприступов. Основными компонентами лечения являются:
- Антиконвульсанты. На первом этапе проводится монотерапия в максимально переносимой дозировке. Поскольку инфантильные спазмы при синдроме Айкарди отличаются стойкостью к противосудорожным препаратам, то приходится переходить к комбинированной схеме терапии, включающей 2-3 препарата.
- Глюкокортикоиды. Показали свою эффективность при внутримышечном введении в течение 1-3 месяцев. В ряде случаев по назначению врача возможна более длительное глюкокортикоидное лечение.
- Реабилитационная терапия. Направлена на поддержание двигательной активности, предотвращение мышечной атрофии, формирования контрактур при спастичности. Лечебные мероприятия включают массаж и специальную гимнастику, разработанные индивидуально с учетом имеющихся аномалий. Детям со средней степенью когнитивного дефицита показаны занятия с психологом, логопедом для максимально возможного развития навыков.
Поскольку синдром Айкарди является генетическим заболеванием с неуточненными этиофакторами генных мутаций, то его специфическая профилактика затруднительна. Возможно проведение общих профилактических мероприятий, направленных на охрану женщины от вредоносных воздействий в период беременности.
1. Синдром Айкарди/ Милованова О.А.// Журнал неврологии и психиатрии им. С.С. Корсакова - 2011. - 111(7).
2. Синдром Айкарди/ Петухова Н.М., Павловец Л.П., Жакашева А.С.// Вестник Алматинского государственного института усовершенствования врачей. - 2010. - 1.
3. Aicardi syndrome: an epidemiologic and clinical study in Norway/ Lund, C., Bjørnvold, M., Tuft, M., Kostov, H., Rosby, O., Selmer, K. K.// Pediatric Neurology. - 2015. - 52(2).
4. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children/ Glasmacher, M. A., Sutton, V. R., Hopkins, B., Eble, T., Lewis, R. A., Park Parsons, D., & Van den Veyver, I. B. // Journal of child neurology. - 2007. - 22(2).
Синдром кошачьего крика, который еще называют синдром Лежена – это достаточно редко встречающееся хромосомное заболевание, которое характеризуется дефектом строения пятой хромосомы. При этом хромосомном дефекте отмечаются различные тяжелые пороки развития внутренних органов и тканей. Дети с синдромом кошачьего крика очень часто страдают от осложнений, которые возникают при дефекте пятой хромосомы.
По статистическим данным, синдром кошачьего крика является не очень распространенной хромосомной патологией. Так, данное заболевание встречается у 1 ребенка на 30-60 тысяч новорожденных. При этом, возникновение синдрома Лежена не зависит от региона или климатических условий. Однако отмечено, что женский пол является фактором риска.
По сравнению, с большинством других наследственных патологий синдром кошачьего крика имеет относительно благоприятный прогноз. При этой патологии дети могут дожить до зрелого возраста при соответствующем медицинском уходе и недопущении развития серьезных осложнений. Однако, у людей с этим недугом невозможна нормальная жизнь без физических и умственных отклонений.
Интересная информация о синдроме:
- Синдром кошачьего крика первоначально обнаружил и описал в середине двадцатого века генетик Джером Лежен, именем которого и был назван этот синдром.
- Это патологическое состояние имеет специфические симптомы, по которым его можно диагностировать у только родившихся деток.
- Данный синдром отличается специфическим криком ребенка (пронзительный и очень громкий), что напоминает мяуканье кошки. При этом недуге наблюдается дефект развития гортанных хрящей.
- При синдроме Лежена у больных отмечается нормальное количество хромосомного материала, что отличает его от других генетических патологий. Наблюдается только небольшой недостаток пятой хромосомы, что и является причиной заболевания.
Причины синдрома кошачьего крика
Синдром Лежена является хромосомной патологией. Ведущая причина синдрома кошачьего крика заключается в изменении структуры хромосом в генной информации ребенка. Геном содержит всю информацию о каком-либо организме. В состав генома входит 23 пары хромосом. Хромосомные заболевания возникают по причине нарушения целостности какой-либо из хромосом.
Синдром кошачьего крика характеризуется тем, что в геноме каждая пятая хромосома будет иметь дефект в любой клетке, независимо от ее функции. При данной патологии пятая хромосома не имеет короткого плеча, на котором локализуется большое количество генов. Такое патологическое состояние в генетике носит название делеции (отсутствие определенного участка ДНК).
Существует несколько вариантов мутаций, которые являются факторами развития синдрома:
- Абсолютное отсутствие короткого плеча на пятой хромосоме. Данный вариант характерен для очень тяжелого течения болезни.
- Уменьшение короткого плеча характеризуется потерей определенной генной информации. При этой форме отмечается потеря генной информации, что приводит к появлению данного синдрома.
- Мозаичный вариант синдрома Лежена является относительно легким течением заболевания (менее выраженные физические и психические отклонения). При данной форме заболевания изначально геном меняет свою структуру во время роста зародыша. Во время деления пятой хромосомы короткое плечо было утеряно, что явилось причиной заболевания.
Синдром кошачьего крика, мутация которого находится в пятой хромосоме, наблюдается при всех вышеперечисленных вариантах отклонений. При этом отмечаются специфические симптомы, которые являются результатом деления клеток, имеющих дефектный ген. Деление таких клеток характеризуется меньшей интенсивностью, так как у них отсутствуют нужные химические компоненты. За счет этого дети с синдромом кошачьего крика зачастую имеют низкую массу тела.
Как правило, дефект в пятой хромосоме наследуется от одного из родителей. Основная причина синдрома кошачьего крика не ясна, так как развитие этой патологии обусловлено множеством факторов, которые воздействуют из окружающей среды. Данные факторы приводят к нарушению процесса деления зиготы в самом начале беременности или повреждению половых клеток одного из родителей.
Факторы, которые провоцируют развитие патологии пятой хромосомы:
- Возраст женщины, которая является матерью ребенка. Вероятность наследственных заболеваний увеличивается с возрастом матери. Для синдрома эта зависимость является довольно слабой. При данной патологии риск развития заболевания увеличивается после 45 лет у матери. При этом возраст отца не влияет на риск развития синдрома Лежена;
- Курение. Курение особенно опасно в молодом возрасте, когда происходит активное формирование репродуктивной системы. Это является частой причиной хромосомной патологии;
- Алкоголь действует таким же образом, как и курение (наблюдается нарушение биохимических процессов, что существенно повышает риск хромосомных аномалий);Некоторые лекарственные средства негативно влияют на репродуктивные органы, в том числе и на мутацию хромосом. Особенно это касается приема препаратов в первом триместре беременности. Это способствует развитию риска мозаичной формы синдрома Лежена. При этом наркотические вещества являются наиболее тератогенными;
- Синдром кошачьего крика, мутация при котором может наблюдаться при наличии внутриутробных инфекций;
- Радиоактивное излучение, направленное на половые органы, часто проявляется хромосомными мутациями;
- Неблагоприятные экологические условия. Ученые отмечают, что в регионах, где добывают токсические полезные ископаемые, часто наблюдается рождение детей с хромосомными болезнями.
Причина синдрома кошачьего крика часто кроется в одном из этих факторов, но встречаются случаи, когда у родителей, на которых не воздействовали вышеперечисленные факторы, рождаются дети с синдромом кошачьего крика.
Внешность новорожденных с синдромом кошачьего крика
Ученые определили комплекс симптомов и синдромов, которые в совокупности характерны для синдрома кошачьего крика. Данные симптомокомплексы можно увидеть сразу после рождения ребенка. Типичные симптомы заболевания, которые наблюдаются сразу после рождения малыша:
- Специфический плач новорожденного малыша.
- Аномалии развития костей черепа.
- Определенная форма глазных щелей.
- Атипичная форма ушных хрящей.
- Агенезия нижней челюсти.
- Низкая масса тела при рождении.
- Аномалии развития костного аппарата кисти.
- Косолапость.
Синдром кошачьего крика постоянно проявляется специфическим плачем ребенка. Дефект пятой хромосомы клинически проявляется в первые минуты после рождения ребенка характерным криком в виде мяуканья кошки. Этот крик по тональности отличается от крика нормальных детей. Причиной этого является:
- Уменьшение размеров хряща надгортанника;
- Уменьшение просвета дыхательных путей в проекции надгортанника;
- Аномальное увеличение эластичности хрящевой ткани;
- Образование складок на слизистой оболочке, которая выстилает хрящи гортани.
Так как эти изменения происходят в области голосовых связок, то у детей изменяется тональность голоса.
Аномалии развития головы
Дети с синдромом кошачьего крика в более, чем в 80 % случаев, имеют аномалии развития формы черепной коробки. Чаще всего наблюдается микроцефалия, которая сопровождается уменьшением размеров черепа. Поэтому у новорожденных голова имеет продолговатую форму и пропорционально она меньше размеров туловища. Для того, что подтвердить диагноз микроцефалии, необходимо сделать краниометрию. Микроцефалия всегда сопровождается энцефалопатией различной степени тяжести.
Специфическая форма глазных щелей
Данный симптом характерен для многих хромосомных заболеваний, в том числе и для синдрома Лежена. В основном эта аномалия обусловлена патологической формой костей черепа. Дети с синдромом кошачьего крика имеют нарушения развития глаз, которые можно отличить по четырем признакам:
- Антимонголоидный глазной разрез отличается тем, что медиальный угол глаза располагается всегда выше наружного;
- Косоглазие характеризуется асимметричным расположением роговиц по отношению к векам;
- Гипертелоризм глаз отличается широкой посадкой глазных яблок;
- Эпикантус представляет собой складку у внутреннего угла глаза.
Нарушение формы наружного уха
Для новорожденных детей с синдромом кошачьего крика характерно наличие изменения в строении и локализации ушных раковин. Наиболее частой патологией является птоз ушных раковин. Это является следствием недоразвития хрящей уха, что визуально представлено уменьшением размеров ушей. Также около ушных раковин на коже могут наблюдаться своеобразные уплотненные кожные узелки.
Агенезия нижней челюсти
Недоразвитие нижней челюсти проявляется микрогнатией или микрогенией. Во время беременности из-за хромосомных аномалий кость, которая формирует нижнюю челюсть, может не достигнуть нужных размеров. Все это приводит к втягиванию подбородка по отношению к верхнечелюстной кости. Частые формы микрогнатии – двухсторонняя или односторонняя. В целом нарушение развития нижней челюсти у таких детей приводит к затруднению при питании (ребенок не может полностью сомкнуть губы около соска, что приводит к нарушению сосательного рефлекса).
Патологически сниженный вес ребенка
Новорожденные дети с синдромом кошачьего крика часто имеют аномально низкий вес при рождении. Это является следствием тяжелых нарушений развития внутренних органов.
Патология развития костей костного аппарата
Также может наблюдаться клиндактилия, которая характеризуется нарушением формы пальцев в суставах кисти и стопы.
Косолапость
Данный признак проявляется при наличии аномалий развития костного аппарата нижней конечности. Косолапость – это нарушение расположения стопы по отношению к оси голени. В будущем у таких деток могут проблемы с ходьбой. Совокупность этих симптомов можно диагностировать на пренатальном этапе.
Особенности детей с синдромом кошачьего крика
Выживаемость детей с данным синдромом достаточно высока, поэтому многие из них достигают подросткового возраста. Дети с синдромом кошачьего крика имеют такие фенотипические особенности внешнего вида:
- Умственная отсталость;
- Снижение мышечного тонуса;
- Сниженная координация движений;
- Запоры;
- Лунообразное лицо;
- Укороченная шея;
- Лабильная нервная система;
- Ухудшение зрения.
Умственная отсталость
Часто данный симптом становится заметным в первые годы жизни ребенка и является одним из важных диагностических признаков данного заболевания на фоне полного здоровья.
Сниженный мышечный тонус
Этот симптом развивается при патологии нервной системы или неполноценном развитии определенных мышц. Клинически это заметно в виде повышенной усталости детей во время ходьбы.
Сниженная координация движений
Данное проявление синдрома кошачьего крика развивается при недоразвитии мозжечка, так как у таких деток наблюдается микроцефалия.
Запоры
Этот симптом отмечается вследствие патологически суженного желудочно-кишечного тракта, а также из-за нарушения нейрогуморальной регуляции кишечника.
Лицо лунообразной формы
Данный симптом возникает в результате нарушения развития костей черепа и долихоцефалии. При этом кости лицевой части черепа больше, чем мозговой.
Укороченная шея
При данном симптоме детям трудно повернуть голову в разные стороны. Это возникает в результате недоразвития шейных позвонков и хрящей между ними.
Лабильность нервной системы
У таких деток часто без обоснованной причины меняется настроение. Это является следствием недоразвития нервной системы. Часто такие дети проявляют повышенную агрессивность и активность в детских коллективах.
Ухудшение зрения
Данная симптоматика возникает из-за нарушения развития органа зрения.
Диагностика синдрома кошачьего крика
Диагностика любой хромосомной аномалии проводится в два этапа. Первый этап заключается в выявлении женщин, у которых повышен риск развития детей с хромосомной патологией. Второй этап проводят для подтверждения определенного заболевания. Таким образом, всем беременным женщинам необходима пренатальная диагностика, которая является комплексом диагностических исследований на дородовом этапе. Благодаря данным мероприятиям на ранних сроках выявляются генетические аномалии, среди которых встречается и синдром кошачьего крика.
Синдром кошачьего крика включает следующие диагностические методы:
- Анамнестические данные;
- Кариотипирование обоих родителей;
- Ультразвуковое исследование;
- Исследование крови на выявление плазменных маркеров;
- Инвазивные методы исследования;
- Исследование на послеродовом этапе.
Детальный сбор анамнеза
Подробный опрос родителей педиатром или генетиком.
Кариотипирование родителей
При повышенном риске развития хромосомной аномалии доктор назначает данное исследование, которое позволяет полноценно изучить структуру клетки и ее ядро.
Частые неспецифические признаки синдрома кошачьего крика на ультразвуковом исследовании:
- Увеличение воротникового пространства;
- Повышенное или сниженное количество околоплодных вод;
- Аномалии развития сердца;
- Брахицефалия или долихоцефалия;
- Непроходимость кишечника;
- Короткие трубчатые кости.
Анализ крови на плазменные маркеры включает такие исследования:
- Определение ХГЧ;
- Выявление уровня протеина А;
- Определение концентрации эстриола;
- Определение альфа-фетопротеина.
Инвазивные методы
Наиболее часто используемые инвазивные методики:
- Кордоцентез;
- Амниоцентез;
- Биопсия хориона.
Послеродовая диагностика включает:
- Консультация врача-неонатолога;
- Функциональное исследование сердца;
- Рентгенография и УЗИ пищеварительного тракта;
- Клинический анализ крови и мочи.
Читайте также:
- Дифференциальная диагностика делирия. Окончательная диагностика делирия.
- Рекрутирование эозинофилов. Общие механизмы рекрутирования эозинофилов.
- Случай гипотиреоза, аутоиммунного тиреоидита и железодефицитной анемии при беременности
- Если вы решили проколоть своей дочурке ушки. Прокалывание ушей
- Внутрисуставные переломы проксимальной фаланги кисти. Диагностика и лечение