Синдром Мак-Кьюна — Олбрайта - лучевая диагностика
Добавил пользователь Владимир З. Обновлено: 16.01.2026
Синдром Мак-Кьюна—Олбрайта—Брайцева (СМОБ) относится к редким формам церебрального преждевременного полового развития (ППР) в сочетании с фиброзной дисплазией костей и асимметричной пигментацией кожи; описан в 1907 г. как болезнь displasia fibrosa polycistia (фиброзная остеодистрофия).
Синдром характеризуется триадой симптомов: фиброзно-кистозной дисплазией костной ткани; асимметричной пигментацией кожных покровов; разнообразными эндокринопатиями, наиболее частой из которых является гонадотропнозависимое преждевременное половое созревание (Петеркова В.А. и др., 1999).
Причину возникновения данного синдрома авторы связывают с мутацией в гене, кодирующем а-субъединицу гуанидинтрифосфат-связывающего белка, принадлежащего к классу стимулирующих (Gsa). Мутация возникает спорадически на постзиготной стадии. Маркером синдрома является пигментация кожи светло-кофейного цвета, неправильной формы, обычно на груди, спине, в области поясницы и бедер.
Эти пятна присутствуют с рождения или распространяются по мере поста ребенка. При обследовании 9 пациенток с клинической картиной СМОБ на молекулярно-генетическом уровне выявлено наличие характерной мутации в участке ДНК, кодирующем молекулу аргинина в 201 положении. Кроме того, при тяжелой и среднетяжелой клинике СМОБ авторам удалось обнаружить мутацию в периферических лейкоцитах — при отсутствии при этом синдроме симптомов со стороны красной крови и кроветворных органов. Авторы предполагают, согласно теории о постзиготном характере мутации при СМОБ, что чем раньше в периоде эмбриогенеза произошла мутация, тем шире распределяется популяция клеток, содержащих мутантную ДНК.
Заболевание встречается только у девочек и представляет врожденную наследственную патологию. Во всей мировой литературе не описано ни одного случая заболевания мальчиков (Сметник В.П., Тумилович Л.Г., 1997). Наиболее тяжелые проявления при этом заболевании — костные нарушения.
Причины развития костной патологии труднообъяснимы, поскольку она развивается на фоне повышения уровня эстрогенов, усиливающих процесс кальцинации костей и костеобразования (Сметник В.П., Тумилович Л.Г., 1997). По мнению S.J. Magalini et al. (1981), фиброзно-кистозная дисплазия — фокальное замещение кортикального слоя пролиферирующими фибробластами, чаще поражаются длинные трубчатые кости. Наличие кист приводит к деформации кости, возникновению патологических переломов с искривлением нижележащего края конечности.
Наиболее типичной является деформация бедра, имеющего вид «пастушьего посоха». Берцовая кость чаще искривляется по вальгус-ному типу. Кроме того, формирование фиброзных кист возможно в костях лицевого скелета и основания черепа. При этом возникают лицевая асимметрия, односторонний экзофтальм, в редких случаях наблюдаются неврологические и психические нарушения (атрофия зрительного нерва, потеря слуха, судороги, задержка психического развития). Процесс патологии костной ткани и частота переломов снижаются после завершения пубертата (Брайцев В.Р., 1954).
Клиника эндокринной патологии проявляется в автономной гиперфункции гипофиза и периферических желез внутренней секреции (Петеркова В.А. и др., 1999).
Среди эндокринных нарушений наиболее частым является преждевременное половое развитие. Последнее начинается в возрасте от 6—9 мес. жизни до 7-летнего возраста и может проявиться в двух формах — неполной и полной. Для полной формы ППР характерно развитие двух половых признаков — менархе и увеличение молочных желез, для неполной — наличие вторичных половых признаков при отсутствии менструаций. Характерным является ускорение роста в длину. По достижении репродуктивного возраста женщины с этой патологией по росту и телосложению не отличаются от женщин, у которых половое развитие началось своевременно. Если при полной форме ППР темп полового развития ускорен, то при неполной он удлинен и иногда превосходит время физиологического развития в 2 раза (Мороз М.Г., 1987).
В отличие от истинных форм ППР при синдроме Мак-Кью-на—Олбрайта—Брайцева не отмечается полового оволосения, диф-ференцировка скелета ускоряется незначительно. Яичники обычно не увеличены и содержат крупные, длительно персистирующие фолликулярные кисты. Уровень гонадотропных гормонов обычно не превышает нормы. Выраженность этой патологии снижается после завершения пубертата. В механизм его вовлечена рецептор-нозависимая аденилат-циклиновая система неспецифической активации, которая стимулирует гормонообразование без воздействия тропных гормонов.
Уровень гонадотропных гормонов в крови, по данным большинства исследователей, не превышает возрастной нормы. Реакция гонадотропных гормонов на введение люлиберина также соответствует допубертатным значениям (Kaufmann M.A. et al., 1986).
Следовательно, данная патология по этиологическим факторам и по патогенезу остается не полностью изученной.
Больные данной группы нуждаются в комплексном обследовании и консультации гинеколога-эндокринолога, лучевого диагноста, эндокринолога, офтальмолога, травматолога, а также в определении уровня тропных и гонадотропных гормонов.
Синдром Мак-Кьюна — Олбрайта - лучевая диагностика
Лучевая диагностика фиброзной дисплазии позвоночника (синдроме МакКьюна-Олбрайта)
а) Терминология:
1. Сокращения:
• Фиброзная дисплазия (ФД)
2. Определения:
• Моноостозная форма: единственный очаг поражения
• Полиостозная форма: множественные очаги, нередко сочетание с нарушениями роста
• Синдром МакКьюна-Олбрайта: полиостозная ФД, преждевременное половое созревание, кожные очаги в виде пятен «кофе с молоком»
б) Визуализация:
1. Общие характеристики:
• Локализация:
о Поражения позвоночника встречаются редко:
- Большинство случаев с поражением позвоночника являются полиостозными формами заболевания
- Дуга позвонка > тело позвонка
2. Рентгенологические данные фиброзной дисплазии позвоночника:
• Рентгенография:
о Веретеновидное утолщение кости
о Истончение кортикального слоя
о Характерно наличие матрикса, напоминающего «матовое стекло»:
- Матрикс, однако, может быть представлен в виде как чисто литического, так и чисто склеротического очага
- Может содержать островки хряща
о Узкая переходная зона ± склерозированный край
о Сколиоз
3. КТ при фиброзной дисплазии позвоночника:
• Структура матрикса и переходная зона видны лучше, чем на рентгенограммах
• Разрушение кортикального слоя встречается нечасто
4. МРТ при фиброзной дисплазии позвоночника:
• Т1-ВИ:
о Низкая или промежуточная интенсивность сигнала
• Т2-ВИ FS:
о Неоднородная промежуточная или высокая интенсивность сигнала
• Т1-ВИ с КУ:
о Вариабельное контрастное усиление
5. Радиоизотопные исследования:
• Костная сцинтиграфия:
о Минимальное или выраженное усиление захвата изотопа
о Выраженность захвата изотопа не коррелирует с клиникой заболевания
6. Рекомендации по визуализации:
• Наиболее оптимальный метод диагностики:
о КТ: толщина среза 1-3 мм
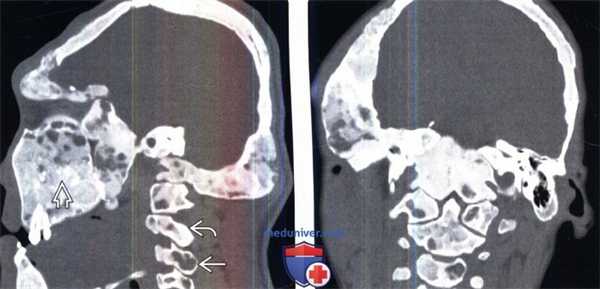
(Слева) Сагиттальный КТ-срез: тяжелый случай полиостозной формы фиброзной дисплазии (ФД) с поражением черепа, лицевого скелета и шейного отдела позвоночника. Некоторые очаги поражения имеют вид «матового стекла», тогда как другие-чисто литические. Также визуализируются отдельные фокусы кальцифицированного хряща.
(Справа) На фронтальном КТ-срезе у этого же пациента не прослеживается трабекулярная структура большинства видимых на срезе костей вследствие замещения трабекул фиброзным матриксом. Истончение компактных пластинок плоских костей черепа является характерным признаком заболевания.
в) Дифференциальная диагностика:
1. Аневризмальная костная киста (АКК):
• Шарообразное увеличение объема дуги позвонка
• Уровни жидкости на КТ и МР-изображениях
• Может сочетаться с фиброзной дисплазией
2. Болезнь Педжета:
• Изменения в литическую фазу процесса могут выглядеть аналогично ФД
• Репаративная фаза отличается другими особенностями: утолщение кортикальных пластинок, грубые утолщенные костные трабекулы, увеличение объема кости
3. Остеобластома:
• Обычно более четко виден костный матрикс
• Может напоминать матрикс в виде «матового стекла»
4. Туберозный склероз:
• Склеротические костные очаги
• Характерные изменения головного мозга
5. Остеосаркома:
• ± аморфный незрелый костный матрикс
• Широкая переходная зона
• Разрушение кортикального слоя, мягкотканный компонент
• Периостальная реакция
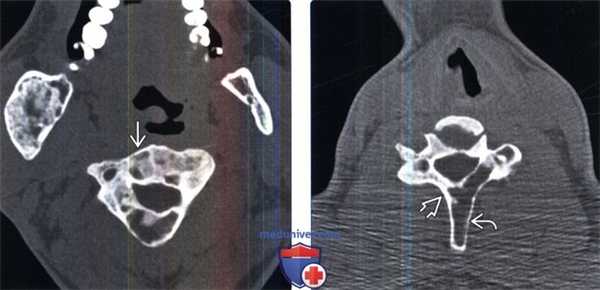
(Слева) Аксиальный КТ-срез: изменения различной плотности в пределах одного позвонка в сочетании с практически полным замещением костного мозга. Подобные изменения типичны для ФД и не должны служить основанием для подозрений на злокачественный характер процесса.
(Справа) Аксиальный КТ-срез: литический очаг ФД в задних элементах позвонка. Наличие узкой переходной зоны (иногда склеротической) помогает отличить ФД от более агрессивных поражений. Тела позвонков обычно поражаются в меньшей степени, чем задние элементы.
г) Патология:
1. Общие характеристики:
• Генетика:
о Спорадическая мутация гена GNAS
• Сочетанные изменения:
о Эндокринные нарушения: гиперпаратиреоз, акромегалия, преждевременное половое созревание, фосфатурия
о Может сочетаться с АКК
2. Макроскопические и хирургическое особенности:
• Белесые или бежевые очаги зернистой структуры
3. Микроскопия:
• Строма образована рыхлой фиброзной тканью
• Небольшие островки новообразованной кости («азбучный суп»)
• Иногда встречаются фокусы хрящевой ткани
д) Клинические особенности. Течение заболевания и прогноз:
• Нарушения роста, патологические переломы часто наблюдаются при полиостозной форме заболевания
• Изредка наблюдается саркоматозная трансформация с развитием остеосаркомы или злокачественной фиброзной гистиоцитомы
е) Диагностическая памятка. Следует учесть:
• На основании данных лучевых методов исследования не следует путать заболевание с болезнью Педжета:
о При болезни Педжета наблюдается утолщение кортикальных пластинок и костных трабекул
о ФД приводит к истончению кортикальных пластинок и замещению костных трабекул фиброзной тканью
Синдром Мак-Кьюна-Олбрайта - клиника, диагностика, лечение
Синонимы: синдром Олбрайта, фиброзная дисплазия полиостотическая.
Определение. Наследственное заболевание, которое проявляется сочетанием нарушения пигментации кожи с фиброзной дисплазией костей и эндокринной патологией.
Историческая справка. Первую публикацию заболевания выполнил A.Weill в 1922 г., описав 9-летнюю девочку с преждевременным половым созреванием, хрупкими костями и кожной пигментацией. В 1932 г. были опубликованы изменения, характерные для этого синдрома (Vera Gaupp). Далее заболевание описали американский педиатр D.J. McCune (Донован Джеймс Мак-Кьюн) в 1936 г. и американский эндокринолог F.Albright с соавторами в 1937 г. Фуллер Олбрайт и соавторы в 1937 г. на основании 21 наблюдения сообщили о системном заболевании, названном ими «синдромом, характеризующимся диссеминированным фиброзным оститом, полями пигментации и эндокринными расстройствами с преждевременным половым созреванием у девочек».
Этиология и патогенез. Причиной развития синдрома являются мутации в гене GNAS1, локализованном в локусе 20q13.2. Ген кодирует гуанин-нуклеотид-связывающий белок (G-протеин). Тип наследования неизвестен. Синдром не передается по наследству. Мутации случайные, они происходят во время внутриутробного развития ребенка.
Частота. Синдром встречается относительно редко (1:100000 и 1:1000000 населения).
Возраст и пол. Изменения кожи чаще обнаруживаются у девочек и, как правило, сразу при рождении.
Поражения кожи. Сразу после рождения или вскоре после него на коже появляются светло-коричневые пятна неправильных очертаний типа географической карты, с изрезанными краями в виде «берега Майна», как правило, крупных размеров. Пигментные пятна нередко располагаются участками и более выражены на пораженной стороне, особенно часто — на задней поверхности шеи, спине, бедрах, в поясничной области.
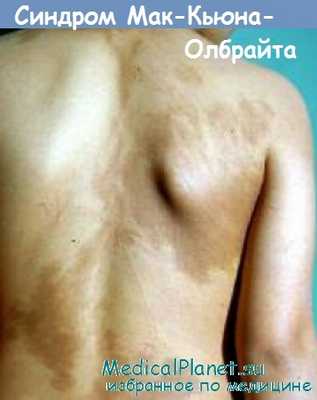
Патология костной ткани. Возникают множественные кистозные очаги остеопороза с участками остеосклероза и гиперостоза (особенно в костях черепа). Это обусловливает патологическую ломкость костей, псевдоартрозы, деформацию костных отверстий, что вызывает неврологическую симптоматику, боль. Длинные кости имеют предрасположенность к искривлению. Деформация черепа и лицевого скелета может быть значительной и сопровождаться снижением зрения и слуха. Поражение костей обычно асимметричное и может быть единственным симптомом заболевания.
Эндокринная патология. У девочек развиваются признаки раннего полового созревания (менструации и вторичные половые признаки часто появляются в возрасте 7 лет). У мальчиков половое развитие нормальное, у них могут быть явления гипергенитализма или атрофии половых желез. У пациентов могут также обнаруживаться гипертиреоз, гиперпаратиреоидизм, гиперпролактинемия, гиперсоматотропизм, синдром Иценко—Кушинга.
Диагноз ставят на основании наличия пятен цвета «кофе с молоком», патологии костной ткани, преждевременного полового развития, гипертиреоза, аномалии надпочечников, акромегалии. При лабораторных исследованиях выявляют повышение уровня гормонов щитовидной железы, паращитовидных желез, надпочечников, а также гормона роста и пролактина. При рентгенологическом исследовании выявляют фиброзную дисплазию, которая затрагивает несколько костей. При МРТ может выявляться аденома гипофиза. При генетическом тестировании обнаруживают ген GNAS1.
Дифференцируют в группе наследственных заболеваний, при которых также обнаруживаются пятна цвета «кофе с молоком». Кофейные пятна не являются достоверным признаком данного синдрома, так как они чаще обнаруживаются у здоровых людей и у пациентов с нейрофиброматозом. Однако у больных с синдромом Мак-Кьюна—Олбрайта пятна цвета «кофе с молоком», как правило, крупные, имеют неправильные очертания, изрезанные края и локализуются преимущественно вблизи костной патологии.
Течение и прогноз. Заболевание клинически неоднородное. Наряду со случаями классической триады признаков, существуют атипичные и неполные формы синдрома. Выраженность симптомов и тяжесть заболевания также сильно изменчивы. С возрастом патология костной ткани прогрессирует. Развитие одной костной патологии по частоте превышает полный симптомокомплекс в 30-40 раз. Злокачественные опухоли встречаются редко (менее 1%). Как правило, обнаруживаются остеосаркомы, особенно на фоне ЛТ, иногда рак щитовидной железы, яичка и молочной железы.
Лечение симптоматическое. Преждевременное половое созревание можно затормозить ингибиторами синтеза эстрогена.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
ИРФ-1 — инсулиноподобный фактор роста 1-го типа
МРТ — магнитно-резонансная томография
СКТ — спиральная компьютерная томография
СТГ — соматотропный гормон
ТМО — твердая мозговая оболочка
Синдром МакКьюна—Олбрайта (McCune—Albright) — это генетически обусловленное заболевание, для которого характерна триада клинических проявлений: 1) эндокринные нарушения, самым частым из которых является преждевременное половое созревание; 2) характерная кожная пигментация по типу пятен «кофе с молоком» (café au lait); 3) фиброзная остеодисплазия.
В российской литературе первым синдром описал В.Р. Брайцев в 1922 г., определив его как «фиброзные опухоли». Спустя 16 лет эндокринолог Ф. Олбрайт сформулировал признаки заболевания как «диссеминированный фиброзный остит, поля пигментации и эндокринные расстройства с преждевременным половым созреванием у девочек».
Причиной заболевания является соматическая мутация гена, кодирующего α-субъединицу G-белка, что приводит к внутриклеточной активации циклического аденозинмонофосфата (ц-АМФ). Клинические проявления зависят от количества и качества мутировавших на эмбриональной стадии клеток. Для диагноза достаточно наличия двух признаков из триады перечисленных клинических симптомов [1, 2]. В ряде случаев у больных с синдромом МакКьюна—Олбрайта могут выявляться различные эндокринные заболевания, в частности аденомы гипофиза. Опухоли гипофиза по гормональной активности оказываются реже кортикотропиномами, чаще — соматотропиномами — «чистыми» или с сопутствующей гиперсекрецией пролактина. Акромегалия у данной группы пациентов обычно сопровождается выраженной гиперсекрецией соматотропного гормона (СТГ) [3].
Изменение костей скелета часто имеет множественный, преимущественно асимметричный, иногда односторонний характер [4]. Нередко фиброзная остеодисплазия протекает бессимптомно с медленно прогрессирующим течением. Поражение костей свода черепа проявляется его выраженной деформацией; костей орбиты — экзофтальмом; костей основания черепа, в основном, — симптомами компрессии черепно-мозговых нервов, в том числе зрительного нерва, структур внутреннего уха [3]. Поражение костей скелета нередко сопровождается патологическими переломами. Менее чем в 1% случаев в очагах остеодисплазии развивается остеосаркома [1, 5].
Больные с синдромом МакКьюна—Олбрайта поступают в нейрохирургическую клинику в основном с аденомой гипофиза, продуцирующей СТГ, либо с аденомой смешанного типа, продуцирующей СТГ и пролактин [2]. Медикаментозная терапия аналогами соматостатина и/или агонистами дофамина в случае их эффективности часто рассматривается как единственный вариант лечения, поскольку возможность безопасного хирургического вмешательства считается ограниченной и даже невозможной ввиду часто выраженного поражения костей основания черепа. Эффективность и безопасность применения лучевой терапии однозначно не определена из-за малого числа наблюдений и потенциального риска саркоматозной трансформации пораженных костей [6, 7]. Появление симптомов компрессии образований хиазмальной области (основной — снижение зрения) определяет показания к нейрохирургическому лечению. При этом транссфеноидальный подход к турецкому седлу считается трудновыполнимым из-за выраженности деформации тела основной кости, сопряженной с полным отсутствием анатомических ориентиров, затрудняющих ориентацию, повышающих риск повреждения внутренней сонной артерии при отклонении оси доступа [8]. Использование нейронавигации считается обязательным для безопасного выполнения этого вмешательства [9].
В настоящее время в ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» более 90% аденом гипофиза оперируют, используя трансназальный эндоскопический доступ. Постоянное улучшение эндоскопической техники, накопление хирургического опыта, разработка современных методов пластики дефектов основания черепа позволяют нам рутинно выполнять самые сложные трансназальные эндоскопические операции [10—13].
Мы представляем результаты трансназального эндоскопического удаления двух соматотропином у пациентов с синдромом МакКьюна—Олбрайта. Основная клиническая информация о пациентах представлена в табл. 1—4. Таблица 1. Основные антропометрические показатели двух пациентов Таблица 2. Гормональный профиль пациентов до и после операции Таблица 3. Динамика зрительных функций пациентов до и после операции Таблица 4. Морфологическая характеристика опухолей
Клинический случай № 1
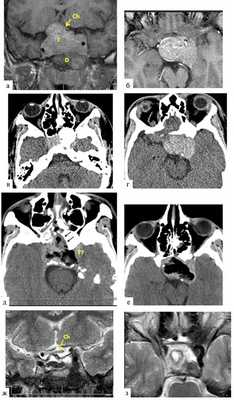
Юноша 14 лет. С раннего возраста опережал сверстников в росте, особенно рост ускорился в последние 3 года, появились симптомы несахарного диабета в виде жажды и полиурии. В 2007 и 2008 гг. у пациента произошли повторные переломы левой локтевой кости на фоне выявленной фиброзной дисплазии левых лучевой и плечевой костей. В ноябре 2015 г. выполнено оперативное вмешательство в связи с юношеским эпифизиолизом головки левой бедренной кости. Поводом для проведения магнитно-резонансной томографии (МРТ) головного мозга в 2015 г., в результате которого обнаружена СТГ-секретирующая аденома гипофиза, послужило снижение зрения (см. табл. 2, 3, рис. 1). Рис. 1. Клинический случай 1. а, б — картина, полученная при МРТ до операции; в, г — картина, полученная при СКТ до операции; д, е — картина, полученная при СКТ после операции; ж, з — картина, полученная при МРТ после операции. Т — опухоль; D — очаг фиброзной остеодисплазии; Ch — хиазма.
При обследовании в Национальном медицинском исследовательском центре нейрохирургии им. акад. Н.Н. Бурденко установлен следующий диагноз: акромегалия, активная фаза; аденома гипофиза; пангипопитуитаризм (вторичный гипотиреоз, гипокортицизм, гипогонадизм); несахарный диабет; синдром МакКьюна—Олбрайта; полиоссальная фиброзная дисплазия; хиазмальный синдром (см. табл. 2, 3).
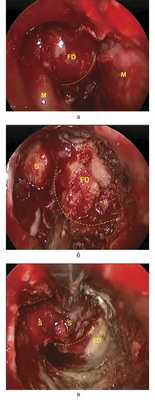
Нами произведено эндоскопическое эндоназальное транссфеноидальное удаление крупной эндо-супра-латеро (S)селлярной опухоли гипофиза. Выполнен двусторонний доступ к основной пазухе. Левая половина пазухи заполнена крупным оссификатом, переходившим на дно седла (рис. 2). Рис. 2. Клинический случай 1. Интраоперационные фотографии. а — кость, пораженная фиброзной остеодисплазией (fd), расположенная перед турецким седлом (м — слизистая полости носа); б — правая половина турецкого седла (s), обнаруженная после частичной резекции пораженной кости (fd); в — обе половины турецкого седла (s) после полной резекции пораженной кости (fd). Бором трепанированы кости дна седла. После рассечения твердой мозговой оболочки (ТМО) обнаружена гетерогенная ткань аденомы гипофиза замазкоподобной консистенции, и для ее удаления потребовались отсосы увеличенного диаметра. Постепенно, разделяя опухоль на фрагменты кюретками, удалось удалить опухоль отсосами практически полностью, в том числе и из латеральных отделов левого кавернозного синуса. Однако тонкие плотные пласты опухоли остались в нескольких местах на ТМО — отделить их не удалось. Из обоих крупных узлов — правого переднего и левого заднего — опухоль удалена. На фоне выведения ликвора в дренаж капсула опухоли расправилась, ликвореи не было. Учитывая, что к концу операции образовалась огромная полость с кровоточащими стенками и после нагрузочных тестов не выявлено ликвореи, решено вход в полость капсулы удаленной опухоли не закрывать.
Гистологическое исследование подтвердило наличие соматотропиномы с высоким уровнем маркера пролиферативной активности опухолевой клетки Ki-67 — 6—7% (см. табл. 4).
Послеоперационный период протекал без особенностей. Профилактически проводилось люмбальное дренирование в течение трех суток. Назальной ликвореи не было. В неврологическом статусе — без отрицательной динамики. После операции отмечено повышение остроты зрения левого глаза (см. табл. 3).
При контрольной спиральной компьютерной томографии (СКТ) выявлена картина практически полного удаления опухоли (см. рис. 1), при этом клинико-биохимической ремиссии акромегалии не было (см. табл. 2). Начата комбинированная терапия пролонгированными аналогами соматостатина и каберголином, на фоне которой сохранялись высокие уровни инсулиноподобного фактора роста 1-го типа (ИРФ-1) и СТГ. С учетом инвазивного характера опухоли и отсутствия ремиссии принято решение о проведении стереотаксической лучевой терапии.
Клинический случай № 2
Мужчина 28 лет. В течение 10 лет отмечал появление изменений внешности: увеличение носа, кистей рук, стоп, губ, языка. Поводом для выполнения МРТ послужило снижение зрения, развивавшееся в течение последних 2 лет. Диагностирована акромегалия, активная фаза (см. табл. 1, 2). Пациенту ранее проводилась терапия аналогами соматостатина, однако эффекта не было.
При обследовании в Национальном медицинском исследовательском центре нейрохирургии им. акад. Н.Н. Бурденко обнаружены аденома гипофиза; акромегалия, активная фаза; хиазмальный синдром; синдром МакКьюна—Олбрайта; полиоссальная фиброзная дисплазия в виде поражения костей свода и основания черепа, кожная пигментация по типу пятен «кофе с молоком». Выявлены также гормонально-неактивные новообразования обоих надпочечников.
Поражение основной кости носило асимметричный характер в виде утолщения ее крыльев и переднего наклоненного отростка справа, правой половины решетчатой кости, ската тела основной кости вплоть до рострума. Полость пазухи основной кости отсутствовала. Нами произведено эндоскопическое эндоназальное транссфеноидальное удаление эндосупраселлярной опухоли гипофиза. С использованием навигации («BrainLab», Германия) различными борами выполнена туннелеобразная трепанация по средней линии в толще резко утолщенной кости, измененной вследствие дисплазии. В результате существенной погрешности навигации по вертикали первоначально удалось выйти на развилку основной артерии. Горизонтально расположенная ТМО над местом обнаружения основной артерии представлялась «дном» турецкого седла, однако при рассечении ТМО обнаружена ткань, более всего напоминавшая мозговое вещество. Одновременно справа от средней линии обнаружен небольшой темный узел ткани, которая, будучи покрытой ТМО, более всего напоминала венозный синус. При рассечении ТМО обнаружена ткань, похожая на опухоль гипофиза, но, учитывая небольшой объем узла и наличие у него костных стенок, поиски турецкого седла решено продолжить. После получения результатов срочной биопсии, подтвердившей, что оба образца тканей являются фрагментами аденомы гипофиза, и после выполнения СКТ, подтвердившей выполнение трепанации строго по средней линии, но непосредственно под дном турецкого седла, трепанация расширена вверх. Обнаружена передняя стенка турецкого седла, из полости которого удалена мягкая синюшного цвета аденома гипофиза. Опухоль распространялась в полость правого кавернозного синуса, где после ее аспирации обнаружено заднее колено сонной артерии. В полость синуса уложена гемостастическая марля, в турецкое седло и на стенки образовавшейся полости уложена гемостатическая губка Тахокомб («Linz», Австрия). На дефект ТМО в области основной артерии уложены лоскут аутофасции, фрагмент жировой ткани, еще один лоскут фасции, костная пластина от носовой перегородки, еще один фрагмент жировой ткани, укрытый мукопериостальным лоскутом. Слои пластики герметизированы фибрин-тромбиновым клеем.
Гистологическое исследование подтвердило наличие соматотропиномы с высоким уровнем Ki-67 5—6% (см. табл. 4). Послеоперационный период протекал без особенностей. Отмечено улучшение зрительных функций (см. табл. 3). При осмотре оториноларингологом назальная ликворея не выявлена.
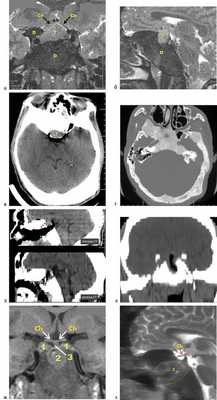
При контрольной МРТ через 3 мес после операции отмечено формирование «пустого турецкого седла». Инфраселлярно в сформированном в утолщенной кости дефекте сохраняется пластический материал. В области обоих кавернозных синусов — небольшие остатки опухоли, что объясняет отсутствие клинико-биохимической ремиссии заболевания (см. табл. 2, см. рис. 3). Рис. 3. Клинический случай 2. а, б — картина, полученная при МРТ до операции; в, г — картина, полученная при СКТ до операции; д — интраоперационная картина СКТ: до (верхнее изображение) и после (нижнее изображение) выполнения дополнительной резекции кости в области передней стенки турецкого седла; е — картина, полученная при СКТ после операции; ж, з — картина, полученная при МРТ после операции. Т или 1 — опухоль; D — очаг фиброзной остеодисплазии; Ch — хиазма; 2 — зона резецированной измененной дисплазией кости; 3 — стебель гипофиза. Пациенту рекомендовано проведение лучевой терапии на область остаточной опухоли.
В наших наблюдениях клинические проявления синдрома МакКьюн—Олбрайта носили классический характер. Размер и расположение аденом гипофиза, вне зависимости от состояния пораженных костей основания черепа, определяли выбор трансназального доступа как оптимального для достижения максимально полного и безопасного удаления. Остеодиспластические изменения основной кости у пациентов были различной степени выраженности. В обоих случаях поражение основания черепа было асимметричным, но в 1-м случае полость основной пазухи и анатомические ориентиры на контралатеральных пораженных костях сохранены, что позволило, ориентируясь по ним, резецировать пораженную кость до получения доступа к турецкому седлу.
Во 2-м случае никаких анатомических ориентиров не сохранилось — резекция кости была возможна только под навигационным контролем. Отсутствие ошибки по горизонтали позволило не потерять ориентир средней линии. Ошибка по вертикали стала причиной того, что на первом этапе операции кость резецирована вплоть до ската ТМО, но под турецким седлом. Только выполнение этапной компьютерной томографии позволило сориентироваться и выполнить доступ к опухоли без интраоперационных осложнений. Для быстрой и безопасной резекции уплотненной кости во 2-м случае потребовалось использовать нетрадиционный для трансназальной хирургии инструмент — пневматическую дрель с борами увеличенного диаметра (5—6 мм).
Технически само удаление ткани опухоли практически не отличалось от стандартной трансназальной операции. Целями удаления гормонально-активной макроаденомы гипофиза являются обеспечение декомпрессии хиазмальной области и достижение гормональной ремиссии заболевания. Нормализация уровня СТГ и ИРФ-1 достигается в 80% случаев после удаления соматотропином [14]. В наших наблюдениях мы не получили желаемого результата. Так, несмотря на рентгенологическую картину почти полного удаления опухоли, не произошло нормализации уровня СТГ и ИРФ-1. В 1-м случае это объясняется выраженной инфильтрацией ТМО плотной опухолью, а во втором случае — распространением опухоли в кавернозные синусы.
Применение нестандартных методик на завершающем этапе операции обеспечило неосложненный послеоперационный период. В 1-м случае оставление открытым входа в значительную по объему полость капсулы опухоли создало условия для максимально быстрой и эффективной декомпрессии зрительных нервов. Отсутствие гемостатиков и пластических материалов в полости капсулы позволило в кратчайшие сроки после удаления опухоли рентгенологически верифицировать ее радикальность и планировать лучевую терапию.
Во 2-м случае выполнение сложной пластики основания черепа предотвратило развитие послеоперационной ликвореи и позволило максимально быстро активизировать пациента.
Медикаментозная терапия аналогами соматостатина обычно малоэффективна у пациентов с синдромом МакКьюна—Олбрайта. Поэтому с учетом инвазивного характера роста опухоли, высокого индекса пролиферативной активности опухолевых клеток и отсутствия ремиссии акромегалии для снижения гиперсекреции гормона роста и предотвращения роста опухоли рекомендована стереотаксическая лучевая терапия, несмотря на данные о возможном риске злокачественной трансформации ткани фиброзной дисплазии на фоне облучения.
Заключение
Костные деформации структур основания черепа при синдроме МакКьюна—Олбрайта могут носить крайне выраженный характер. Тем не менее при установлении диагноза аденомы гипофиза у подобных пациентов выполнение трансназальной операции возможно. Технически это очень сложная задача и она может быть выполнена только высококвалифицированными нейрохирургами, использующими современную навигационную технику, а также нестандартные методики проведения операции.
Участие авторов:
Концепция и дизайн исследования — М.А., П.Л., Л.И., Н.А.
Сбор и обработка материала — М.А., П.Л., Л.И., Н.А., Д.С., Л.А., Е.И., А.М.
Написание текста — М.А., Д.С.,
Редактирование — П.Л., Л.И., Н.А.
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflict of interest.
Комментарий
В статье описаны два редких наблюдения эндоскопического трансназального удаления соматотропных аденом гипофиза у пациентов с синдромом МакКьюна—Олбрайта. Оба случая интересны наличием у больных деформации и фиброзных разрастаний костных структур основания черепа, сложностью выполнения хирургического вмешательства и их нестандартными завершениями. Содержание работы полностью соответствует тематике журнала. Научная новизна и практическая значимость, безусловно, высокие, поскольку хирургическое лечение пациентов с этой патологией ранее было чрезвычайно травматичным или невозможным. Появление эндоскопических технологий, навигационного оборудования и опытных в лечении данной патологии нейрохирургов позволило успешно провести эти вмешательства. Дизайн статьи выбран в стиле описания клинических случаев с последовательным изложением протокола операции, результатов и послеоперационного состояния пациентов с решением вопросов о дальнейшей тактике лечения. Список литературы представлен достаточно полно. Резюме кратко характеризует содержание работы, имеются все необходимые разделы и список ключевых слов. Работа произвела на меня очень благоприятное впечатление. Лаконичность изложения, информативность рисунков и упорядоченность клинических данных в виде таблиц делают восприятие материала легким. Работа будет интересна как нейрохирургам, так и ряду специалистов смежных областей.
Синдром Олбрайта

Синдром Мак-Кьюна-Олбрайта является гетерогенным заболеванием, которое независимо друг от друга описали американский педиатр D. J. McCune и эндокринолог F. Albright в 1936-1937 гг. Заболевание редкое, врожденное, но не наследственное, сопровождается метаболическими нарушениями, признаками преждевременного полового развития и деформаций скелета. Первые клинические проявления отмечаются в раннем детстве или подростковом возрасте в виде нарушений развития, полиоссальной фиброзной дисплазии костей, кожной пигментации по типу «кофе с молоком» и эндокринных аномалий, включая тиреотоксикоз, синдром Кушинга, акромегалию.
Приблизительно в 10 раз чаще диагностируют синдром Олбрайта Мак-Кьюна у девочек, чем у мальчиков. Википедия отмечает, что частота встречаемости в человеческой популяции — 1 на 100 тыс. или на миллион особ.
Патогенез
В результате постзиготной мутации гена GNAS1, участвующего в клеточном сигнализировании белков GS-α , обеспечивающих сопряжение рецепторов лютеинизирующих и фоликолостимулирующих гормонов с молекулами аденилатциклазы в цитологических структурах яичников. Постоянное влияние мутантного белка активизирует аденилатциклазу и повышение внутриклеточного цАМФ, а также стимулирует усиление секреции стероидных женских половых гормонов фолликулярными эстрогенпродуцирующими кистами, вызывающими преждевременное половое развитие даже при отсутствии гонадотропинов. Такая же картина наблюдается и «автономное производство» наблюдается с гормонами щитовидной железы, кортизолом и гормонами роста. Прогрессирование патологии вызвано образованием клеток, уже содержащих мутантные белки.
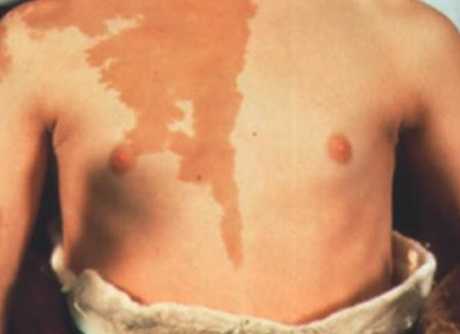
Пигментация при синдроме Олбрайта
Кроме того, рост уровня цАМФ приводит к увеличению продукции меланина и образованию лентигиозных пятен, очаговой гиперпигментации на фоне нормальных значений АКТГ и меланотропина.
Фиброзно-кистозная дисплазия в результате усиленной кальцинации под действием эстрогенов осуществляется путем замещения нормальных костных структур и образованных кальцинатов фиброзными массами, при этом нарушается равновесие процессов резорбции и регенерации костей, пролиферации мезенхимальных стромальных клеток. Наблюдается формирование очагов деструкций и псевдоопухолей.
Классификация
Болезнь Олбрайта бывает разной, в зависимости от тяжести течения различают:
- скрытое заболевание – без каких-либо признаков костной или эндокринной патологии, а также без симптоматики необычной пигментации;
- неполные формы синдрома, например, в процесс патогенеза вовлечения костная ткань, возможны эндокринные изменения и псевдогипопаратиреоз, но нет кожных проявлений;
- псевдогипопаратиреозили наследственная остеодистрофия Олбрайта — заболевание, которое также поражает костные ткани, но механизм и этиология отличаются — патология вызвана нарушением резистентности к паратгормону, метаболизма кальция и фосфора; псевдогипопаратиреоз также проявляется в детском возрасте и выражается в виде низкого роста, круглого лица, умственной отсталости, ожирения, гипотиреоза, гипогонадизма, сахарного диабета и пр.
Причины
Синдром Олбрайта Маккьюна возникает в результате генетических нарушений на ранних этапах эмбриогенеза. До сих пор не установлен тип наследования, все описанные случаи были спорадические.
Симптомы
Клиническая картина заболевания проявляется в виде целого симпатокомплекса, состоящего из:
- преждевременного гонадотропиннезависимого полового развития, развивающегося позже и медленнее нежели при других формах ранней половозрелости;
- пятнистой пигментации кожи – лентиго, пятна с четкими границами светло-коричневого цвета обычно есть от рождения на груди, спине пояснице, бедрах и в местах костных деформаций;
- эндокринных расстройствах, вызывающих тахикардию, необъяснимую утрату веса, глазные симптомы гипертиреоидизма;
- полиоссальной фиброзной остеодисплазии, мышечной слабости и остеопороза, вовлекающей в процесс чаще длинные кости, нарушения проявляются в виде изуродованных черт лица, более вытянутом телосложении, искривленных и асимметричных, укороченных запястьях, что затрудняет движения во время ходьбы, вызывает хромоту, сильные боли и повышает вероятность патологических переломов.

Первые признаки преждевременной половой зрелости начинаются с маточных кровотечений, не имеющих цикличного характера и вызваны кратковременными повышениями концентраций эстрогенов и гонадотропных гормонов в кровяном русле. Такие колебания провоцируют образования кист в яичниках. У детей в возрасте 5-10 лет наблюдается ранний рост груди и гинекомастия. Её нежная этиология не приводит к формированию вторичных половых признаков — Синдром Мак-Кьюна-Олбрайта-Брайцева не вызывает раннего полового оволосения или дифференцировки фигуры. Истинное половое созревание возможно после окончания пубертатного периода.
Анализы и диагностика
При подозрении на синдром Мак Кьюна Олбрайт проводят:
- рентгенологические исследования, которые выявляют псевдокисты, сегментарное поражение любых отделов скелета – чаще нижних конечностей, в более редких случаях — верхнего плечевого пояса и черепа;
- УЗИ брюшной полости позволяет обнаружить кисты яичников;
- лабораторные исследования для выявления повышенного уровня щелочной фосфатазы в кровотоке, сниженного количества гонадотропинов – в анализах мочи и повышенной экскреции лютеинизирующего гормона, 17-гидрокси и 17-оксикортикостероидов.
Лечение
Лечение назначают симптоматическое, оно имеет свои сложности в зависимости от индивидуальных особенностей и клинической картины. Аналоги гонадолиберина не оказывают терапевтического эффекта, так как процесс секреции эстрогенов спровоцирован не избытком гонадотропинов. Для купирования процесса гонадального стероидогенеза у особ мужского пола эффективным может оказываться кетоконазол и андрокур. При лечении медроксипрогестерона ацетатом возможно развитие гипокальциемии, поэтому, если процесс развития ранней половозрелости сопровождается тяжёлыми множественными поражениями костных тканей, то терапию необходимо назначать с осторожностью.
Для угнетения гиперэстрогенизиции и удлинения скелета могут быть назначены препараты тестостерона. Чтобы противодействовать дисплазии костей следует вводить:
- бисфосфонаты;
- ингибиторы антагонистов остеокластов.
Больным необходимо регулярно, каждые 3 мес. посещать эндокринолога для коррекции проблем с щитовидной и другими железами.
Читайте также:
- Смешенные опухоли пищевода. Доброкачественные опухоли и изменения желудка.
- Причины и признаки воспаления пищевода
- Дифференциальная диагностика острого аппендицита. Болезни с которыми дифференцируется аппендицит
- Клеточные и субклеточные механизмы регуляции пролиферативных процессов при воспалении
- Бородавчатый туберкулез кожи. Скрофулодерма - колликвативный туберкулез кожи