Синдром Марфана. Деформация Маделунга, болезнь Лери — Вейля
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) - редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птицеголовость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефалия» принадлежат Н.Зеске в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Синдром Секкеля - редкий синдром, характеризующийся тяжелой низкорослостью, низким весом при рождении, очень маленькой головой (микроцефалия), большим лбом, большими глазами, низкими ушами, большим носом и небольшим подбородком. Дефекты костей рук и ног, вывихи локтевых суставов и бедер, неспособность выпрямлять колени и крипторхизм, также часто встречаются у детей с этим синдромом.
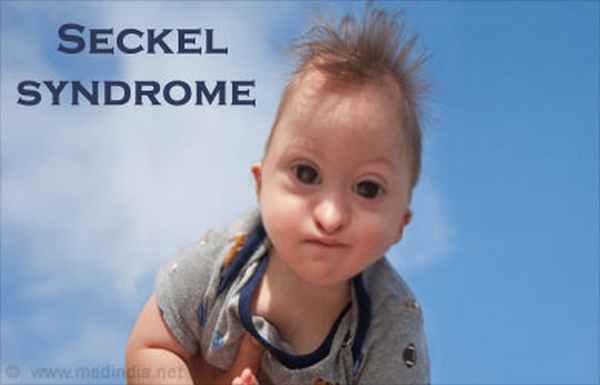
Хромосомная нестабильность и недопроизводство всех типов клеток крови (панцитопения) отмечаются у некоторых пациентов. Это заболевание является генетическим. Оно наследуется по аутосомно-рецессивному типу. Синдром Секкеля является неоднородным генетическим заболеванием, которое может быть связано с мутациями в генах, расположенных на хромосомах 3 и 18.
Следует отличать синдрос Секкеля от другиз заболеваний с низкорослостью. Для этого проводится дифференциальная диагностика при характерных дополнительных симптомах.
Низкая масса тела при рождении в случае доношенной беременности, внутриутробная задержка роста. Этот частый симптом свидетельствует о наличии генетически обусловленного снижения потенции роста тканей, недостаточности плаценты или же нарушений роста во внутриутробном периоде токсического или воспалительного генеза. Если отсутствует дополнительная симптоматика и вместе с тем имеющуюся клиническую картину нельзя отнести к какому-либо определенному заболеванию, говорят о примордиальной низкорослости. При отсутствии других симптомов последняя описывается как семейное заболевание.
В спорадических случаях следует думать о необратимом внутриутробном повреждении редупликационного потенциала клеток неизвестным патогенным фактором. При чисто примордиальной низкорослости дифференцировка скелета соответствует хронологическому возрасту.
- а) Примордиальная низкорослость: синдром Рассела - Сильвера.
- б) Синдром Секкеля: гетерогенная группа примордиальных карликов с микроцефалией, большим носом, скошенным подбородком и интеллектуальным недоразвитием.
- в) Прогерия (синдром Гатчинсона-Джилфорда): замедление роста и развитие начиная со 2-3-го года жизни, преждевременное старение, атрофия кожи, выпадение волос.
- г) Синдром Блума: телеангиэктатическая эритема щек, долихоцефалия.
- д) Синдром Корнелии де Ланге; умственное недоразвитие, густые, сросшиеся над переносицей брови, микроцефалия, усиленное оволосение тела.
- е) Лепрехаунизм (синдром Донохуа): отсутствие или недоразвитие подкожной жировой клетчатки, гипертрофия клитора или полового члена, усиленное оволосение.
- ж) Синдром Фанкони: панцитопения, гипоплазия I пальца руки или нарушение его закладки, пигментные аномалии.
- з) Семейная, или полигенная, низкорослость, при которой в большинстве случаев наблюдается только легкая задержка роста во внутриутробном периоде.
- и) Трисомия 18.
Очень короткие конечности (увеличенное отношение верхнего сегмента к нижнему). К этой группе прежде всего относятся заболевания скелета, обусловленные поражением эпифизов и метафизов. При дифференциальной диагностике обязательно следует подумать об атиреозе или тяжелом гипотиреозе (особенно учитывая возможности их лечения). Фосфатный диабет также может сопровождаться легкой диспропорциональной низкорослостью с преобладанием верхнего сегмента. Пропорции могут существенно меняться в процессе роста. Здесь представлены сведения о пациентах старшего детского и юношеского возраста в соответствии с обобщением Спрангера.
- а) Ахондроплазия (хондродистрофия): относительно большая голова с провалившимся основанием носа, особенное укорочение предплечий и голеней, кифоз, типичные рентгенологические находки.
- б) Гипохондроплазия: слабо выраженная диспропорциональная низкорослость, переразгибание суставов, люмбальный лордоз, типичные рентгенологические данные.
- в) Псевдоахондроплазия: пропорции, как и при ахондроплазии, но с нормальным формированием мозгового и лицевого черепа, относительно позднее по сравнению с ахондроплазией проявление заболевания, отсутствие типичных рентгенологических симптомов со стороны таза.
- г) Синдром Эллиса-Ван-Кревельда: порок сердца, дистрофия ногтей и пальцев стоп, эписпадия.
- д) Синдром Маркезани: изменения хрусталиков, как при синдроме Марфана (дрожание хрусталиков, может быть шаровидный хрусталик), брахидактилия, возможны легкие контрактуры.
- е) Метафизарная хондродисплазия, тип Шмидта: укорочение трубчатых костей (больше проксимальных), грибовидно вздутые эпифизы с размытыми очертаниями.
- ж) Метафизарная хондродисплазия, тип Мак-Кусика: редкие тонкие волосы, брови и ресницы, переразгибание суставов, типичная рентгенологическая картина.
- з) Метафизарная хондродисплазия, тип Енсена: утолщенные суставы с костным ограничением подвижности, формирование косолапости стоп, типичная рентгенологическая картина.
- и) Дисхондростеоз (синдром Лери - Вейля): укорочение предплечья и лучевой кости по отношению к локтевой, дорсальный, поддающийся редрессации подвывих дистального конца локтевой кости (деформация Маделунга).
- к) Пикнодизостоз: короткие концевые фаланги, большой мозговой череп, большие роднички (которые могут быть открытыми до зрелого возраста), спонтанные переломы.
- л) Карликовость с микромелией: гипоплазированные локтевая и малоберцовая кости, а также нижняя челюсть.
- м) Карликовость с микромелией: характерная "треугольная" деформация большеберцовой кости.
- н) Псевдогипопаратиреоз: ожирение, обращающее на себя внимание круглое лицо, укороченные пальцы рук, изменения метаболизма.
- о) Псевдопсевдогипопаратиреоз.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
Дисхондростеоз Лери-Вейля
Дисхондростеоз Лери-Вейля – наследственное заболевание, характеризующееся развитием многочисленных скелетных аномалий, которые в совокупности приводят к карликовости и другим нарушениям. Симптомами этого состояния являются низкий рост, мезомелия (укорочение средних участков конечностей), деформации костей предплечий, голени и запястья, сколиоз. Диагностика дисхондростеоза Лери-Вейля производится на основании данных осмотра больного, изучения его наследственного анамнеза, рентгенологических исследований скелета и молекулярно-генетических анализов. Специфического лечения заболевания не существует, применяют симптоматическую терапию, включая хирургическую коррекцию деформированных и укороченных костей.
Общие сведения
Дисхондростеоз Лери-Вейля или энхондральный политопный дизостоз – генетическая патология, обусловленная нарушением формирования энхондральных костей. Вызывает карликовость и многочисленные скелетные аномалии. Это состояние было впервые описано в 1929-м году французскими врачами неврологом А. Лери и педиатром Ж. Вейлем, результаты исследований в 1959-м году подтвердили их коллеги и соотечественники М. Лами и К. Биненфельд. Долгое время считалось, что данная патология является аутосомно-доминантной, однако гены, ответственные за ее развитие, найти не удавалось. На современном этапе развития генетики причину этого заболевания обнаружили в половых хромосомах. При этом дисхондростеоз Лери-Вейля не является сцепленным с полом заболеванием, поскольку гены, дефекты которых приводят к его развитию, расположены на так называемых псевдоаутосомных участках половых хромосом, а наследование этих участков не зависит от пола больного. По этой причине заболевание поражает лиц обоих полов, однако фенотипически оно тяжелее протекает у женщин. Дисхондростеоз Лери-Вейля является достаточно редким состоянием, его точная встречаемость не выяснена.
Причины дисхондростеоза Лери-Вейля
Дисхондростеоз Лери-Вейля является генетическим заболеванием с псевдоаутосомно-доминантным механизмом наследования. Гены, мутации которых приводят к этому состоянию, расположены на половых хромосомах (как X, так и Y), но передаются потомству как обычные аутосомные участки. Этими генами являются SHOX и SHOXY, располагающиеся на коротких плечах X и Y хромосом (точнее, на псевдоаутосомном участке 1 или PAR1). Продуктом экспрессии данных генов становятся одноименные белки, которые представляют собой высокоактивные транскрипционные факторы – наибольшее их количество у здорового человека выявляется в костной ткани. Дисхондростеоз Лери-Вейля развивается по причине нарушения структуры или экспрессии протеинов SHOX и SHOXY, что влечет за собой искажение процессов деления и дифференцировки клеток в энхондральных костях (позвонках, длинных трубчатых костях конечностей).
Для развития дисхондростеоза Лери-Вейля мутация генов SHOX или SHOXY обязательно должна быть в гетерозиготном состоянии – у гомозигот с такими генетическими дефектами возникает намного более тяжелое заболевание (мезомелическая дисплазия Лангера). В некоторых случаях при наличии характерных фенотипических проявлений данной патологии мутации в генах SHOX и SHOXY могут не выявляться. Чаще всего генетический дефект при этом локализуется на высококонсервативных некодирующих участках, которые располагаются до и после данных генов. Эти участки контролируют процессы транскрипции и экспрессии SHOX, то есть выступают его энхансерами. При их повреждении процессы экспрессии вышеуказанных генов нарушаются, что также приводит к развитию дисхондростеоза Лери-Вейля. Аналогичными проявлениями обладают и дупликации последовательностей данных генов. Иногда наличие мутаций в гене SHOX приводит не к развитию таких серьезных скелетных аномалий, а к состоянию, известному как идиопатический низкий рост.
Симптомы дисхондростеоза Лери-Вейля
В большинстве случаев дисхондростеоз Лери-Вейля можно диагностировать уже при рождении ребенка, однако описаны случаи возникновения выраженных симптомов заболевания и у детей старшего (вплоть до подросткового) возраста. У новорожденного характерным признаком патологии является укорочение верхних и нижних конечностей относительно общих пропорций тела. В дальнейшем к этому проявлению добавляются варусные деформации лучевой и большеберцовой кости во фронтальной плоскости и локтевой – в сагиттальной. Нарушается структура костей и их соединений в области запястья, что также приводит к деформациям (болезнь Маделунга) и затрудняет движения кисти. Скорость роста больных дисхондростеозом Лери-Вейля замедлена, в конечном итоге длина тела взрослых людей с таким состоянием не превышает 130 сантиметров.
Довольно часто при дисхондростеозе Лери-Вейля у пациентов регистрируются различные искривления позвоночника, главным образом – сколиоз. При этом аномалий костей черепа, ребер и таза не наблюдается. Пороки развития внутренних органов для этого заболевания также нехарактерны, интеллект больных полностью сохранен. Не страдают и функции половой системы, практически все больные способны иметь детей – лишь в тяжелых случаях у женщин вынашивание ребенка может быть затруднено из-за вторичных нарушений (тяжелого сколиоза или других скелетных аномалий). Таким образом, дисхондростеоз Лери-Вейля проявляется только аномалиями позвоночника и длинных трубчатых костей конечностей, не затрагивая другие системы и остальные части скелета.
Диагностика и лечение дисхондростеоза Лери-Вейля
Диагностика дисхондростеоза Лери-Вейля производится на основании изучения статуса пациента, рентгенографии костей конечностей и позвоночника, генетического анализа. При осмотре обращают на себя внимание укороченные конечности – руки в области предплечий и ноги за счет голеней. Нередко невооруженным взглядом можно увидеть деформацию этих элементов скелета. Важным проявлением дисхондростеоза Лери-Вейля являются затрудненные супинация и разгибание кисти за счет деформации запястья по типу Маделунга. Всегда отмечаются низкорослость больных и недостаточность их общего физического развития.
При рентгенографии выявляются укорочение и деформации локтевых, лучевых и большеберцовых костей, в некоторых случаях обнаруживается аплазия малоберцовой кости, приводящая к выраженной деформации стопы. Межкостный промежуток предплечья сильно расширен, кости запястья деформированы, их первый ряд формирует изгиб, который визуально как бы раздвигает дистальные концы локтевых и лучевых костей. На рентгенограммах при дисхондростеозе Лери-Вейля также может выявляться сколиоз позвоночного столба различной степени выраженности. Врачом-генетиком может быть произведена генетическая диагностика этого заболевания – секвенирование последовательности генов SHOX и SHOXY для выявления точковых мутаций, анализ их количества для исключения дупликации, изучение структуры энхансеров. Дифференциальную диагностику дисхондростеоза Лери-Вейля следует производить с другими типами скелетных дисплазий.
Специфического лечения дисхондростеоза Лери-Вейля не существует, симптоматическая терапия заключается в снижении нагрузки на кости и суставы, хирургической коррекции резко выраженных деформаций и аномалий. Возможно использование так называемой корригирующей остеотомии. При сильном нарушении структуры стоп (что наблюдается при аплазии малоберцовой кости), обусловленном дисхондростеозом, прибегают к артроризу. Иногда по желанию больного хирургическими методами производят удлинение конечностей путем наложения дистракционных аппаратов после продольной остеотомии. При тяжелых формах сколиоза может потребоваться помощь хирурга-ортопеда и вертебролога.
Прогноз и профилактика дисхондростеоза Лери-Вейля
Несмотря на то, что дисхондростеоз Лери-Вейля часто приводит к инвалидизации больного, а значительно укороченные конечности сильно снижают качество его жизни, прогноз заболевания относительно выживаемости благоприятный. Это связано с тем, что нарушение затрагивает только костные элементы конечностей и отчасти позвоночника, не влияя на внутренние органы, интеллектуальное развитие и обмен веществ. При тяжелом сколиозе могут развиваться вторичные нарушения дыхательной и сердечно-сосудистой систем, поэтому данное осложнение дисхондростеоза Лери-Вейля требует своевременного лечения. Профилактика этого состояния производится только методом пренатальной генетической диагностики, что особенно актуально при наличии подобного заболевания у родственников.
Синдром Марфана. Деформация Маделунга, болезнь Лери — Вейля
Синдром Марфана. Деформация Маделунга, болезнь Лери — Вейля
Изредка с синдромом Марфана сочетается глухота. Мы подозреваем, что у больных, описанных Brock и Bucklers, была гомоцистннурия. Ganther обнаружил глухоту, но приписал се наблюдавшимся у больного повторным инфекциям среднего уха. У глухих больных, представленных Lloyd, не был достаточно убедительно доказан синдром Марфана. Kelemen изучил височную кость 11-месячного ребенка с установленным диагнозом синдрома Марфана. Из наружного отверстия ненормально суженного водопровода преддверия выпячивалась костная губа. В улитке круглое окно было выпячено в сторону лестницы, утрикуло-ампулярнос пространство и эндолимфатический ход были расширены. Возможно, многие из таких больных имели синдром кератоконуса, миопии, марфаноидной внешности и нейросенсорной глухоты.
Прогрессирующая оссифицирующая фиброплазия (прогрессирующий оссифицирующий миозит). Lutwak, Ludman с сотр. и Letts описали глухоту у больных с прогрессирующей оссифицирующей фиброплазией, заболеванием соединительной ткани, передающуюся аутосомио-доминантным геном с низкой пепетрантно-стью (McRusick). Обычно отмечается микродактилия 2 пальцев рук, ног и прогрессирующий анкилоз шейного отдела позвоночника, позже начинается окостенение парасиипальных мышц, мышц плечевого и тазового поясов. Глухота в одних случаях нейросснсорного типа, в других —проводящего (Ludman et al., Letts). Причина глухоты неизвестна. Нарушение слуха, очевидно, не является постоянным симптомом болезни.
Ключично-черепная дисплазия (ключично-черепной дизостоз). Синдром, характеризующийся гипоплазией или отсутствием ключиц и различными другими скелетными аномалиями (червеобразные косточки, позднее закрытие черепных швов, родничков и симфиза, непрорезавшиеся и дополнительные зубы и т. д.), наследуется по аутосомно-доминантному типу. Изредка этот синдром сочетается с прогрессирующей проводящей или смешанной глухотой и концентрическим сужением наружного слухового прохода (Nager, DeReynier, Davis, Gay, Jaffee, Fons). Клетки сосцевидного отростка отсутствуют. Исследование вестибулярной системы, проведенное Gay, выявило редуцированную реакцию на калорическую стимуляцию, но аналогичное исследование Fons патологии не обнаружило. Томография продемонстрировала деформацию слуховых косточек (Fons).
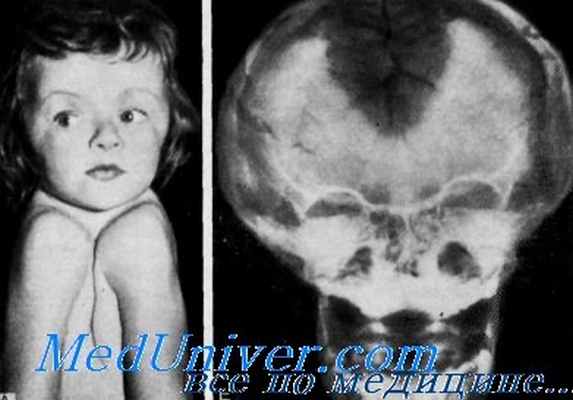
Ключично-черепная дисплазия
Дисхондростеоз (деформация Маделунга, болезнь Лери — Вейля). Дисхондростеоз характеризуется деформацией дистальных отделов лучевой и локтевой костей и проксимальных отделов запястья, а также мезомелической карликовостью (Herdman et al.).
Nassif и Harboyan описали братьев с двусторонней проводящей глухотой на уровне до 40—50 дБ. Наружный слуховой проход был узким. Молоточек отсутствовал, а рудиментарная наковальня не была связана с деформированным стременем. В одном ухе было невозможно определить chorda tympani.
Семейная остеома среднего уха. Остеомы среднего уха встречаются чрезвычайно редко. Thomas описал 2 случая: у 10-летнего мальчика и у его 6-летней сестры. Родители и 2 старших сибса были здоровы. Исследование среднего уха у мальчика обнаружило гладкую с широким основанием остеому области пирамиды. Опухоль рспро-странялась и начинала срастаться с суставом между наковальней и стременем. Барабанная перепонка была нормальной и слух, согласно аудиометрическим данным, тоже был нормальным. У сестры была обнаружена односторонняя остеома меньших размеров. Ее слух, низкий до операции, после операции восстановился до нормы. У обоих сибсов наблюдался экссудативный отит. Предполагали, что причиной его было костное новообразование в среднем ухе.
Ombredanne описал подобную опухоль в среднем ухе у 57-летнего мужчины, страдавшего в течение 20 лет двусторонней прогрессирующей глухотой. Другие члены семьи не были поражены й данных о первичном экссудативном отите не имелось.
Синдром Марфана
Синдром Марфана — это наследуемое заболевание соединительной ткани, характеризующееся патологическими изменениями сердца и сосудов, опорно-двигательного аппарата и глаз. Синдром Марфана выявляется у 1 на 3000—5000 человек, однако есть несколько других наследуемых заболеваний соединительной ткани, которые имеют сходные клинические проявления и похожие опасные осложнения, что делает проблемы, связанные с синдромом Марфана весьма актуальными.
Причиной развития синдрома являются дефекты (мутации) гена фибриллина, одного из важных компонентов соединительной ткани. В результате таких мутаций значительно увеличивается количество специального белка, который приводит к возникновению характерных для синдрома Марфана изменений соединительной ткани.
Синдром Марфана может быть унаследован от одного из родителей или же, (примерно в четверти случаев) быть результатом спонтанной мутации. О спонтанной мутации говорят в тех случаях, когда ранее в семье этим заболеванием никто не страдал. Вероятность наследования синдрома Марфана от больного родителя 50:50.
У некоторых пациентов типичные признаки синдрома Марфана выявляются еще в детском возрасте, но чаще этот диагноз устанавливается гораздо позднее.
Типичными признаками синдрома Марфана являются:
— Со стороны сердечно-сосудистой системы
- Расширение (аневризма) аорты, основного сосуда, который несет кровь от сердца к органам
- Расхождение слоев аорты, что может привести к ее разрыву – расслоению аорты и требует оперативного лечения
- Пролапс митрального клапана
— Со стороны опорно-двигательной системы
- Высокий рост
- Длинные конечности
- Искривление позвоночника (кифоз или сколиоз)
- Деформация грудной клетки
- Длинные пальцы
- Плоскостопие
- Неправильный рост зубов
— Со стороны глаз
- Миопия (близорукость) тяжелой степени
- Подвывих хрусталика
- Отслоение сетчатки
- Ранняя глаукома и катаракта
- Стрии на коже
- Спонтанный пневмоторакс (спадание легкого)
Все перечисленные признаки могут встречаться в разных сочетаниях и диагноз «синдром Марфана» или одного из близких наследственных заболеваний может быть поставлен по совокупности клинических признаков, называемых «Гентскими критериями». В случае выявления одного из внешних признаков, нередко сопутствующих синдрому Марфана следует выполнить следующие инструментальные исследования:
- Электрокардиограмма
- ЭхоКГ – помогает оценить функцию сердца и клапанного аппарата, выявить аневризму восходящей аорты, пролапс митрального клапана
- Осмотр у офтальмолога – позволяет выявить подвывих хрусталика, типичного для данного заболевания
- КТ или МРТ – позволяет выявить проблемы позвоночника, часто встречающиеся у пациентов с синдромом Марфана
Почему пациенту с подозрением на синдром Марфана важно обратиться к врачу?
Наиболее типичным и нередко серьезным осложнением синдрома Марфана и других близких ему наследственных заболеваний соединительной ткани является разрыв или расслоение аорты. Благодаря проведению профилактических мероприятий, а при необходимости выполнении хирургического вмешательства за последние 30 лет в цивилизованных странах удалось существенно снизить опасность этих осложнений и существенно увеличить продолжительность жизни таких больных. Вот почему такие больные должны находится под постоянным врачебным наблюдением, принимать медикаментозные средства, снижающие риск развития серьезных осложнений, выполнять рекомендации, позволяющие уменьшить проявления деформаций скелета и скорректировать зрение.
Синдром Марфана
Синдром Марфана наследуется по аутосомно-доминантному признаку Аутосомно-доминантные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения . Основной молекулярный дефект – результат мутаций в гене, кодирующем гликопротеин фибриллин-1 (FBN1), который является основным компонентом микрофибрилл и помогает клеткам прикрепляться к внеклеточному матриксу. Основной структурный дефект связан с сердечно-сосудистой системой, опорно-двигательным аппаратом и зрительной системой. Также поражаются легочная система и центральная нервная система. Существует много проявлений генетической мутации, вызывающей синдром Марфана; однако распознается она, как правило, по сочетанию длинных конечностей, аневризмы корня аорты и дислоцированных хрусталиков.
Симптомы и признаки синдрома Марфана
Сердечно-сосудистая система
Основные признаки включают:
Самые тяжелые осложнения развиваются в результате патологических изменений корня аорты и восходящей аорты. Интима аорты повреждается преимущественно в местах, подверженных наибольшей гемодинамической нагрузке. Аорта постепенно расширяется или происходит спонтанный разрыв коронарного синуса, иногда в возрасте до 10 лет. Корень аорты расширяется у 50% детей и у 60–80% взрослых, что может привести к аортальной недостаточности Аортальная регургитация Аортальная недостаточность (АН) – это неполное закрытие аортального клапана, приводящее к оттоку крови из аорты в левый желудочек во время диастолы. Причины включают дегенерацию клапанов и расширение. Прочитайте дополнительные сведения , в этом случае прослушивается диастолический шум над клапаном аорты.
Растяжение створок клапанов и сухожильных хорд может привести к пролапсу митрального клапана или его недостаточности; пролапс митрального клапана Пролапс митрального клапана (ПМК) Пролапс митрального клапана (ПМК) – это прогибание створок клапана в полость левого предсердия во время систолы. Наиболее распространенная причина – идиопатическая миксоматозная дегенерация. Прочитайте дополнительные сведенияОпорно-двигательный аппарат
Серьезность симптомов значительно варьируется. Рост пациентов выше средних показателей для возраста и семьи; размах рук превышает высоту. Заметны арахнодактилия; часто с симптомом большого пальца кисти (дистальная фаланга большого пальца выступает за край сжатого кулака). Деформации грудины – килевидная грудная клетка (смещение наружу) или воронкообразная грудь (смещение внутрь) – являются распространенными, как и гиперподвижность суставов (но, как правило, незначительные сгибательные контрактуры локтевого сустава), перегиб колена (выгнутое кзади колено), плоскостопие Плоскостопие (плоские стопы) Эквиноварусная косолапость характеризуется сгибанием подошвы, наклоном пятки внутрь (от средней линии ноги) и приведением переднего отдела стопы (медиальное отклонение от вертикальной оси ноги). Прочитайте дополнительные сведения и паховые грыжи Паховая грыжа у новорожденных Паховые грыжи чаще всего развиваются у новорожденных мальчиков, особенно у недоношенных (частота возникновения около 10%). Наиболее часто паховая грыжа возникает справа, и только в 10% встречается. Прочитайте дополнительные сведения . Подкожно-жировой слой развит недостаточно. Нёбо часто с высоким сводом.
На рисунке показан типичный габитус тела у подростка с синдромом Марфана, включая: кифосколиоз, воронкообразную деформацию грудной клетки и выгнутое назад колено.
С разрешения издателя. Из Macro R: Atlas of Heart Diseases: Congenital Heart Disease . Edited by E Braunwald (series editor) and RM Freedom. Philadelphia, Current Medicine, 1997.
Этот пациент с синдромом Марфана выше, чем члены его семьи, и размах его рук превышает его высоту.
© Springer Science+Business Media
На этой фотографии у человека с синдромом Марфана большой палец кисти выступает за пределы сжатого кулака.
MEDICAL PHOTO NHS LOTHIAN/SCIENCE PHOTO LIBRARY
Синдром Марфана характеризуется аномально длинными пальцами. На этой фотографии видно, что, когда женщина охватывает пальцами запястье другой руки, ее большой палец перекрывает указательный.
Фото предоставлено David D. Sherry, MD.
Зрительная система
Симптомы включают эктопию хрусталика (подвывих или смещение хрусталика вверх) и иридодонез (дрожание радужной оболочки глаза). Края дислоцированных хрусталиков часто можно увидеть через нерасширенные зрачки. Возможна значительная близорукость, а также может возникнуть спонтанное отслоение сетчатки.
Дыхательная система
Могут развиться кистозная болезнь легких и рецидивирующий спонтанный пневмоторакс. Эти нарушения могут вызвать боль и одышку.
Центральная нервная система
Часто выявляют дуральную эктазию (расширение дурального мешка, окружающего спинной мозг), преимущественно в пояснично-крестцовом отделе позвоночника. Это нарушение может вызвать головную боль, боли в нижних отделах спины или неврологические нарушения, проявляющиеся слабостью кишечника или мочевого пузыря.
Диагностика синдрома Марфана
эхокардиография/МРТ (исследование корня аорты, обнаружение клапанного пролапса);
исследование со щелевой лампой (аномалии хрусталика);
Рентген скелетной системы (рук, позвоночника, таза, груди, ног и черепа на характерные аномалии);
МРТ пояснично-крестцового отдела позвоночника (дуральная эктазия)
Диагностика синдрома Марфана может быть затруднена, потому что у многих пациентов наблюдаются лишь некоторые характерные симптомы и признаки, а специфичные гистологические и биохимические изменения отсутствуют. Из-за этой изменчивости диагностические критерии основаны на сочетании клинических данных, семейного и генетического анамнеза. (Более подробную информацию о диагнозе, см. revised Ghent nosology). Тем не менее, диагноз неясен во многих частных случаях синдрома Марфана.
Гомоцистинурия Классическая гомоцистинурия Ряд дефектов в метаболизме метионина приводит к накоплению гомоцистеина (и димера гомоцистеина) с неблагоприятными эффектами, включая тенденцию к тромбообразованию, смещению хрусталика, а также. Прочитайте дополнительные сведения может частично имитировать синдром Марфана, но может быть дифференцирована при обнаружении гомоцистеина в моче. Генетическое исследование мутации FBN1 может помочь установить диагноз у людей, не соответствующих всем клиническим критериям, тем не менее, встречаются случаи негативного результата мутации гена FBN1. Пренатальная диагностика путем анализа мутаций FBN1 гена затруднена вследствие слабой корреляции генотип/фенотип (описано > 1700 различных мутаций).
Стандартную визуализацию скелетной, сердечно-сосудистой и зрительной систем проводят для выявления любых клинически значимых структурных аномалий как имеющих безусловную ценность для постановки диагноза (например, эхокардиография для выявления расширения корня аорты).
В дополнение к критериям, выявленным в системах органов, семейный анамнез (родственников 1-го колена с синдромом Марфана) и генетический анамнез (наличие FBN1 мутации, вызывающей синдром Марфана) считают основными критериями.
Прогноз при синдроме Марфана
Достижения в области терапии и регулярного мониторинга улучшили качество жизни и снизили смертность. Средняя ожидаемая продолжительность жизни увеличилась с 48 лет в 1972 году до почти нормальной, у людей получающих соответствующую медицинскую помощь. Тем не менее, продолжительность жизни среднестатистического пациента по-прежнему ниже, в первую очередь, из-за сердечных и сосудистых осложнений. Эта сниженная продолжительность жизни может оказывать эмоциональную нагрузку на подростков и семьи.
Лечение синдрома Марфана
Индуцирование преждевременного полового созревания у высоких девушек
Элективное восстановление аорты и клапанов
Фиксация и хирургическое вмешательство при сколиозе
Лечение синдрома Марфана направлено на профилактику и лечение осложнений.
У очень высоких девушек индукция преждевременного полового созревания в возрасте 10 лет эстрогенами и прогестероном может уменьшить возможный рост во взрослом возрасте.
Все пациенты должны регулярно получать бета-блокаторы (например, атенолол, пропранолол), чтобы помочь предотвратить сердечно-сосудистые осложнения. Эти препараты снижают сократимость миокарда и пульсовое давление, уменьшают прогрессирование аневризмы корня аорты и риск ее расслоения. Также возможно применение блокаторов рецепторов к ангиотензину II.
Профилактическое хирургическое вмешательство предлагается, если диаметр аорты > 5 см (у детей меньше). Беременные женщины особенно подвержены высокому риску осложнений со стороны аорты; необходимо обсудить рекомендуемую аортальную реконструкцию до зачатия. Тяжелая клапанная недостаточность также восстанавливается хирургически. Профилактика бактериального эндокардита Профилактика Инфекционный эндокардит (ИЭ) – инфекция эндокарда, обычно бактериальная (чаще стрептококковая или стафилококковая) либо грибковая. Он может проявляться лихорадкой, шумами в сердце, петехиями. Прочитайте дополнительные сведения Ведение сколиоза Лечение Идиопатический сколиоз – боковое искривление позвоночника. Диагноз устанавливают на основании клинических признаков, в том числе по результатам рентгенографии брюшной полости. Методы лечения. Прочитайте дополнительные сведенияОценку сердечно-сосудистой, скелетной системы, а также органа зрения (в том числе, эхокардиографию) следует проводить ежегодно. Показано соответствующее генетическое консультирование.
Основные положения
Причиной синдрома Марфана является аутосомно-доминантная мутация гена, кодирующего гликопротеин фибриллин-1, который является основным компонентом микрофибрилл, вследствие чего возможны многочисленные деформации и дефекты.
Проявления варьируют в широких пределах, но основные структурные дефекты затрагивают сердечно-сосудистую и опорно-двигательную системы, а также орган зрения, в результате чего типичным является сочетание удлиненных конечностей, расширения корня аорты и дислокации хрусталиков.
Расслоение аорты является наиболее опасным осложнением.
Диагностика основывается на клинических критериях; часто проводится генетическое исследование.
Для выявления структурных аномалий необходимо провести диагностическую визуализацию органов скелетной и сердечно-сосудистой систем и органа зрения.
Всем пациентам следует назначить бета-блокаторы для профилактики аортальных осложнений; лечение других осложнений следует проводить по мере их возникновения.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
2010 Revised Ghent Nosology for Marfan syndrome: Diagnostic criteria for Marfan syndrome, including a diagnostic calculator
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: