Синдром Меккеля—Грубера - клиника, диагностика
Добавил пользователь Владимир З. Обновлено: 28.01.2026
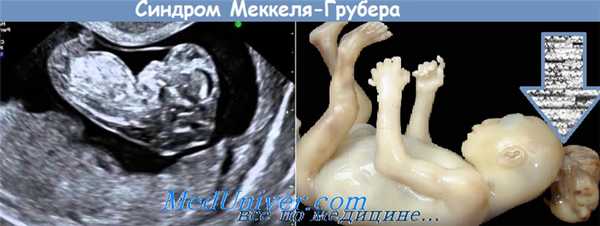
Cиндром Меккеля-Грубера является редкой летальной врожденной множественной аномалией, характеризующейся триадой симптомов: пороки развития мозга (главным образом затылочное энцефалоцеле), увеличенные поликистозные почки и полидактилия, а также сопутствующие аномалии, включая заячью губу/небо, пороки сердца и генитальные дефекты, пороки развития центральной нервной системы, фиброз печени и костные дисплазии.
Распространенность синдрома Меккеля-Грубера в Европе оценивается порядка 1/50 000 рождений. Во всем мире распространенность варьирует от 1/13250 до 1 / 140,000 новорожденных. Распространенность синдрома Меккеля-Грубера значительно выше в финской популяции (1/9000), в Бельгии, среди кувейтских бедуинов (1/3500) и в популяции гуджаратских индусов (1/1300).
Синдром Меккеля-Грубера наследуется по аутосомно-рецессивному типу.
В основе синдрома Меккеля-Грубера лежит дефект цилиарной биологии клетки. Мутации в 14 генах, ответственных за формирование ресничек, были идентифицированы в связи с этим расстройством, часто в контексте близкородственных браков. Большинство из этих генов также ответственны за развитие синдрома Жубера. Возможно, что синдром Меккеля-Грубера является летальным вариантом фенотипа синдрома Жубера.
Плод с синдромом Меккеля-Грубера в лучшем случае выживает в течение от нескольких дней до нескольких недель после рождения или умирает в утробе матери. Основные симптомы со стороны ЦНС включают затылочное энцефалоцеле, гидроцефалию, анэнцефалию, голопрозэнцефалию, а также мальформацию Денди-Уокера. Увеличенные поликистозные почки с кистозной дисплазией - являются обязательным симптомом синдрома Меккеля. Часто наблюдается дисгенез и фиброз печени. Полидактилия может обнаруживаться у всех четырех конечностей плода, как правило, постаксиальная (80%) или редко преаксиальная полидактилия. Отмечается легочная гипоплазия, как следствие маловодия. Может наблюдаться расщелины губы и нёба, микрофтальмия и микрогнатия. Сердечные пороки развития могут включать в себя дефект межпредсердной перегородки, коарктацию аорты, открытый артериальный проток и клапанный стеноз легочной артерии. Присутствует часто неполное развитие внутренних и наружных половых органов, а также крипторхизм у мальчиков.
Диагноз может быть сформулирован на основе УЗИ плода, которое демонстрирует затылочное энцефалоцеле и дисплазию почек. Наличие двух из трех основных пороков развития или наличие двух других аномалий наряду с одной классической особенностью - является достаточными для постановки диагноза синдрома Меккеля-Грубера. Расстройство часто обнаруживается до 14 недели беременности. Молекулярно-генетическое тестирование также может быть использовано для подтверждения диагноза.
Пренатальная диагностика возможна с помощью УЗИ плода, начиная с 12 недель беременности. Могут быть обнаружены энцефалоцеле, кистозные почки и полидактилия.
Никакого лечения синдрома Меккеля в настоящее время не разработано.
Плод с синдромом Меккеля-Грубера умирает в утробе матери или в очень раннем неонатальном периоде от легочной гипоплазии и почечной недостаточности.
Cиндром Меккеля-Грубера наследуется по аутосомно-рецессивному типу, пары имеют 25% риск рецидива.
Пренатальная диагностика синдрома Меккеля-Грубера
Московский областной НИИ акушерства и гинекологии, Москва¹.
Кафедра акушерства и гинекологии N1 Ростовского государственного медицинского университета, Ростов-на-Дону².
УЗИ сканер HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Меккеля-Грубера (дизэнцефалия спланхнокистозная) - комплекс множественных летальных врожденных пороков развития с аутосомнорецессивным типом наследования. Картировано несколько локусов синдрома: тип 1 (ген MKS1 [249000 - регистрационный номер по каталогу моногенных заболеваний OMIM] на хромосоме 17q23), тип 2 (ген MKS2 [603194] на 11q13), тип 3 (ген MKS3 [607361] на 8q), тип 4 (ген MKS4 [611134] на 12q21.3) [1].
Опорными признаками синдрома ("ядром") являются: затылочная черепномозговая грыжа (80%), полидактилия (75%), обычно постаксиальная (чаще поражаются кисти), а также дисплазия почек (95%) [2]. В 15% случаев полидактилия сочетается с синдактилией. Могут наблюдаться камптодактилия, клинодактилия V, косолапость и поперечная ладонная складка.
Для диагностики синдрома, по мнению Г.И. Лазюка и соавт., достаточно наличия любых двух опорных признаков [3]. Кроме ключевых признаков синдрома, возможен широкий спектр сочетанных аномалий: микроцефалия (32%), гидроцефалия (25%), гипо- или аплазия мозолистого тела (13%), аринэнцефалия (17%), гипо- или аплазия мозжечка (28%), в единичных случаях анэнцефалия, гидроцефалия, голопрозэнцефалия, мальформация Dandy - Walker, септальные дефекты сердца, омфалоцеле, кистофиброз печени, расщелины лица и позвоночника. Встречаются также скошенный лоб, низко расположенные деформированные ушные раковины, микрогения, гипертелоризм, реже - гипотелоризм, капиллярные гемангиомы на лбу, зубы новорожденного, липоматозные образования на боковых поверхностях языка. В 25% случаев имеются пороки развития глаз: микрофтальмия (19,2%), реже - анофтальмия, колобомы, катаракта [2, 4].
Частота синдрома Меккеля-Грубера точно неизвестна. По данным B. Auber и соавт. [5], распространенность синдрома в Германии оценивается как 1:135000, в Израиле 1:50000, в Минске - 1:65000 родов [6]. В Финляндии и Татарстане это заболевание встречается необычно часто и достигает 1,1 на 10000 родов. Имеются данные, что оно составляет 5% всех дефектов нервной трубки [7].
В связи с большой редкостью этого синдрома, единичными публикациями в отечественной литературе [6], посвященными его диагностике, представляем собственный опыт пренатальной диагностики 3 случаев синдрома Меккеля-Грубера, каждый из которых был интересен и уникален в плане постановки диагноза.
Наблюдение 1
Беременная А., 32 лет. Данная беременность 3-я, в анамнезе 1 неосложненные роды в срок и 1 артифициальный аборт. Наследственность не отягощена. Мужу 36 лет, здоров. Брак не родственный, супруги производственных вредностей не имеют. Пациентка при сроке гестации 13 недель 3 дня обратилась для проведения скрининговой эхографии в городской перинатальный центр (Ростов-на-Дону). Биохимический скрининг I триместра не проводился. Исследование выполнялось на ультразвуковом сканере с использованием режима поверхностной объемной реконструкции 3D/4D.
При эхографии в срок 13 недель 3 дня фетометрические параметры плода соответствовали гестационной норме, толщина воротникового пространства составила 1,5 мм; длина костей носа - по 2,5 мм. При трансвагинальном сканировании были обнаружены: затылочное энцефалоцеле размерами 7,4х3,5х3,7 мм (рис. 1), полидактилия кистей (рис. 2), увеличенные кистозно-измененные почки: правая - 28,5х21,3 мм; левая - 26,4х18,7 мм (рис. 3). Объем амниотической полости был нормальным для данного срока.
Синдром Меккеля—Грубера - клиника, диагностика
УЗИ, МРТ при синдроме Меккеля-Грубера у плода
а) Определения:
• Синдром впервые описан Иоганном Меккелем в 1822 г. на примере пары сиблингов
• В 1934 г. Георг Грубер описал случаи спланхнокистозной дизэнцефалии у плодов
• Классическая триада признаков:
о Кистозная дисплазия почек (95-100%)
о Энцефалоцеле или другая патология ЦНС (90%)
о Постаксиальная полидактилия (55-70%)
б) Лучевая диагностика:
1. Общие сведения:
• Критерии диагностики:
о Наличие по меньшей мере 2 из 3 классических признаков у плода с нормальным кариотипом
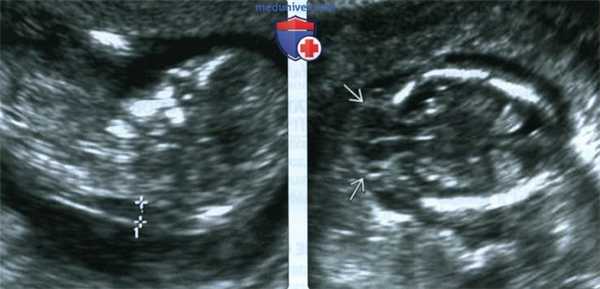
(Слева) ТАУЗИ плода в I триместре. Определяется умеренное утолщение воротникового пространства (кали-перы). По поводу неровных контуров задней части свода черепа выполнено ТВУЗИ.
(Справа) ТВУЗИ подтверждает наличие дефекта задней части свода черепа, а также затылочного энцефалоцеле крупных размеров, содержащее мозжечок целиком. Показан тщательный поиск других пороков развития.
2. УЗИ при синдроме Меккеля-Грубера у плода:
• Мочеполовая система:
о Наиболее устойчивый признак - кистозная дисплазия почек
о Ультразвуковая структура почек варьирует:
- Чаще всего почки значительно увеличены и гиперэхогенны
- Размеры почек увеличены в 10-20 раз
- Могут определяться крупные кисты
о ОЖ может быть значительно увеличена
о Агенезия почки (редко)
о Возможно уменьшение или отсутствие визуализации мочевого пузыря
о Маловодие во II триместре:
- Нередко определяется ангидрамнион
- До тех пор, пока почки не являются основным источником продукции околоплодных вод (I триместр), объем врд соответствует норме
• ЦНС:
о Затылочное энцефалоцеле (60-80%)
о Мальформация Денди-Уокера
о Нередко - микроцефалия
о АМТ
о Вентрикуломегалия
о ГПЭ
• Конечности:
о Постаксиальная полидактилия:
- Добавочный палец может быть уменьшен или искривлен
- Обычно поражены все 4 конечности, однако данный признак является наименее устойчивым
- Визуализация на фоне маловодия может быть затруднена
о Преаксиальная полидактилия встречается реже
о Характерна косолапость
о Укорочение конечностей
о Искривление трубчатых костей
• Пороки развития лица:
о Расщелина верхней губы и нёба
о Микрогнатия
о Микрофтальм
о Пороки развития ушной раковины
о Скошенный лоб
• Сердце:
о Дефекты перегородок
о Коарктация аорты
• Другие пороки развития:
о Фиброз печени:
- Часто обнаруживают при аутопсии
- Диагностика на стадии внутриутробного развития затруднена
- Если заболевание впервые обнаружено в III триместре, находят гепатомегалию и снижение внутрипеченочного кровотока
о Крипторхизм
о Гениталии промежуточного типа
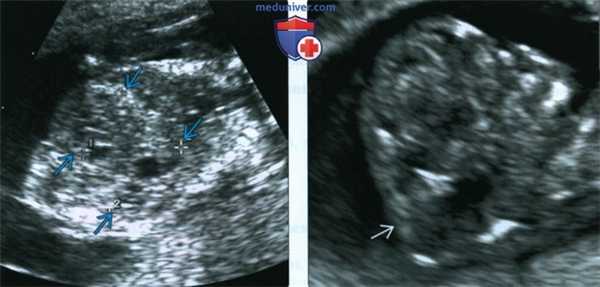
(Слева) Тот же плод. УЗИ брюшной полости, фронтальная плоскость. С обеих сторон определяются увеличенные кистозно-измененные почки. Необходимо помнить, что объем амниотической жидкости в I триместре может быть в норме, даже несмотря на тяжелое двустороннее поражение почек.
(Справа) Во время скринингового УЗИ плода в I триместре, выполненного во время последующей беременности у той же женщины, обнаружено энцефалоцеле. Синдром Меккеля-Грубера имеет аутосомно-рецессивный тип наследования, риск повторного возникновения составляет 25%.
в) Дифференциальная диагностика синдрома Меккеля-Грубера у плода:
1. Т13:
• Многие признаки совпадают
• Аномалии почек (50% случаев):
о Кистозная дисплазия:
- Гиперэхогенные почки, содержащие кисты
- Почки могут быть увеличены, но, как правило, в меньшей степени, чем при синдроме Меккеля-Грубера
о Гидронефроз
• Аномалии ЦНС:
о Секвенция ГПЭ (40%)
о Сообщается о возможном наличии энцефалоцеле, однако значительно реже
• Пороки развития конечностей:
о Постаксиальная полидактилия (75%)
о «Стопа-качалка»
• Маловодие менее характерно:
о Возможно многоводие
• Пороки сердца (80%):
о Дефекты перегородок
о Гипоплазия левых отделов сердца
о Атрезия аортального или митрального клапана
• ЗРП
• Омфалоцеле
2. Синдром Смита-Лемли-Опица:
• Аномалии ЦНС:
о Микроцефалия, ГПЭ, гидроцефалия, АМТ
• Пороки сердца:
о Дефект АВ-канала, межжелудочковой перегородки, гипоплазия левых отделов сердца
• Мочеполовая система:
о Гениталии промежуточного типа, кистозная болезнь почек
• Постаксиальная полидактилия
• Проявления тяжелых форм синдрома совпадают с таковыми синдрома Меккеля-Грубера
• Характерные черты лица:
о Гипертелоризм, короткий вздернутый нос, низкорасположенные уши, микрогнатия, широкий и высокий лоб, эпикантус
3. АРПКП:
• Увеличенные гиперэхогенные почки
• Полидактилия и энцефалоцеле отсутствуют
• Выраженность маловодия варьирует
4. Двусторонняя МКДП:
• Остальные признаки синдрома Меккеля-Грубера отсутствуют
5. Гидролетальный синдром (синдром Салонен-Херва-Норио):
• Полидактилия (часто с удвоением I пальца стопы), гидроцефалия, пороки сердца
• Кистозное поражение почек отсутствует
6. Синдром Барде-Бидля:
• Полидактилия, прогрессирующая дистрофия почек, пороки развития печени, туловищное ожирение
• Энцефалоцеле отсутствует
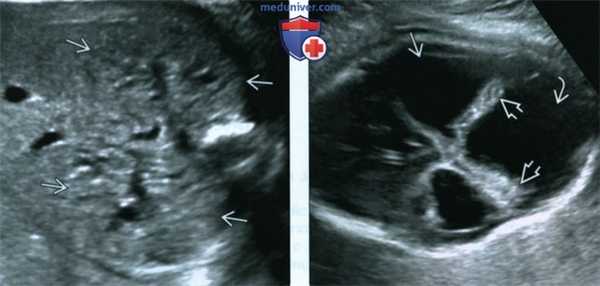
(Слева) УЗИ плода в III триместре, поперечная плоскость, косой срез. Околоплодные воды отсутствуют, определяются увеличенные гиперэхогенные почки.
(Справа) Тот же плод. УЗИ головного мозга. Определяются признаки мальформации Денди-Уокера: расхождение полушарий мозжечка, отсутствие червя, наличие крупной кисты в ЗЧЯ и вентрикуломегалия. Несмотря на то что характерной находкой для синдрома Меккеля-Грубера является затылочное энцефалоцеле, возможны и другие аномалии ЦНС.
г) Патологоанатомические особенности:
1. Общие сведения:
• Генетические факторы:
о Летальная цилиопатия, характеризующаяся генетической гетерогенностью
о Аутосомно-рецессивный тип наследования:
- Риск повторного возникновения - 25%
о Установлено как минимум 11 генов, остальные являются предметом исследований
- MKS1, ТМЕМ216 (MKS2), ТМЕМ67 (MKS3), СЕР290 (MKS4), RPGRIP1L (MKS5), CC2D2A (MKS6), NPHP3 (MKS7), TCTN2 (MKS8), B9D1 (MKS9), B9D2 (MKS10), ТMEМ231 (MKS11)
- Фенотипическая вариабельность объясняется вовлечением нескольких различных хромосом
2. Гистологические особенности синдрома Меккеля-Грубера у плода:
• Почки:
о Кистозная дисплазия почек
о Тяжелый дефицит нефронов
о Кортико-медуллярная дифференциация снижена или отсутствует
о Почки могут быть увеличены в 10-20 раз
• В печени и почках находят миофибробласты
• Фиброз печени (нарушение формирования дуктальной пластинки):
о Недоразвитие внутрипеченочных желчных протоков:
- Реактивная пролиферация желчных протоков
- Дилатация желчных протоков
о Перипортальный фиброз:
- Приводит к облитерации воротной вены
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Большинство случаев могут быть диагностированы в I триместре:
- 90% случаев диагностируются к 14,3±2,6 нед.
о Маловодие во II и III триместрах
• Другие субъективные и объективные симптомы:
о Акушерский анамнез может быть отягощен по данному синдрому
о Повышение уровня а-фетопротеина сыворотки крови матери вследствие энцефалоцеле:
- Энцефалоцеле, покрытое оболочкой, может не приводить к повышению уровня α-фетопротеина
о Широкая фенотипическая вариабельность:
- Сопутствующие нарушения в каждом конкретном случае существенно различаются
2. Демографические особенности:
• 2,6:100 000 родов; частота варьирует в зависимости от региона мира:
о Бельгия - 1:3000
о Финляндия - 1:9000
• М = Ж
• У 5% плодов с энцефалоцеле имеется синдром Меккеля-Грубера
3. Естественное течение и прогноз:
• Заболевание летально:
о Маловодие приводит к гипоплазии легких
о Большинство детей умирают внутриутробно или в течение нескольких часов после рождения
4. Лечение синдрома Меккеля-Грубера у плода:
• Кариотипирование для исключения Т13
• Предлагают прерывание беременности
• В случае сохранения беременности избегают наблюдения за состоянием плода и выполнения КС
• Увеличенная ОЖ может стать причиной дистоции живота
• Для подтверждения диагноза наружный осмотр и аутопсию проводят квалифицированные патологоанатом и генетик
• Для планирования последующих беременностей показано генетическое консультирование:
о Риск повторного возникновения - 25%
е) Особенности диагностики:
1. Важно знать:
• Если визуализация анатомических структур затруднена вследствие маловодия, показана МРТ
• При обнаружении одного из классических признаков синдрома выполняют тщательный поиск других
2. Признаки, учитываемые при интерпретации изображений:
• Лучевые признаки синдрома во многом совпадают с таковыми Т13:
о Для определения кариотипа и консультирования по поводу будущих беременностей выполняют амниоцентез:
- Риск повторного возникновения Т13 составляет 1%, синдрома Меккеля-Грубера - 25%
• Строение почек варьирует от увеличения размеров и гиперэхогенности до полного замещения паренхимы крупными кистами:
о Гигантские размеры почек часто приводят к увеличению ОЖ
Редактор: Искандер Милевски. Дата обновления публикации: 19.11.2021
Синдром Меккеля. Диагностика и прогноз при синдроме Меккеля
Это редко встречающийся летальный синдром, который характеризуется затылочным цефалоцеле, постаксиальной полидактилией и кистозной дисплазией почек. Он может сочетаться со многими другими состояниями, наиболее распространенным из которых является фиброз печени.
Синонимы. Дисэнцефалия с кистозными изменениями внутренних органов и синдром Меккеля (Meckel) (принят в англоязычной литературе). Синдром Меккеля (Meckel) является более предпочтительным названием заболевания и используется как при поиске в интернет-базе данных Medline, так и в Британской энциклопедии врожденных пороков (Birth Defect Encyclopedia). Другими названиями являются синдром Грубера (Gruber) (используется в европейской литературе) и синдром Меккеля-Грубера (Meckel-Gruber).
Распространенность. Точно неизвестна, однако по общепринятому мнению это очень редко встречающаяся патология. По данным D. Bergsma, распространенность синдрома Меккеля (Meckel) составляет 0,2 на 10 000 новорожденных. R. Salonen и R. Norio установили, что частота данного синдрома при рождении варьирует от 0,07 до 0,7 на 10 000 родов.
Генетические нарушения. Локус, ответственный за возникновение синдрома Меккеля (Meckel), локализован на длинном плече хромосомы 17q2.1-q2.4. Фенотипическая вариабельность и случаи без подтвержденной связи с хромосомой 17q предполагают наличие некоторой степени локусной гетерогенности.
Диагностика. В 1981 году F.C. Fraser и A. Lytwyn предположили, что кистозная дисплазия почек является постоянной аномалией при синдроме Меккеля (Meckel), и поэтому для установления диагноза она должна обнаруживаться в сочетании с не менее чем двумя меньшими дефектами. Данная концепция продолжает обсуждаться, при этом распространенность аномалий почек при данном синдроме составляет от 95 до 100%. Почки первоначально имеют микроскопические кисты, которые развиваются, разрушая ее паренхиму и вызывая увеличение органа в 10-20 раз.
Первым эхографическим признаком в большинстве случаев является маловодие вследствие дисфункции почек, возникающее в начале второго триместра беременности, когда продукция мочи должна замещать внеклеточную диффузию в качестве основного источника околоплодных вод. Однако в некоторых случаях синдрома может обнаруживаться нормальное количество амниотической жидкости, и, таким образом, ее наличие не исключает диагноза. В ряде ситуаций мочевой пузырь не будет визуализироваться. Отсутствие эхографических признаков заболевания на ранних сроках у беременной из группы риска по рецидиву заболевания у плода не исключает синдрома Меккеля (Meckel), и в этих случаях рекомендуется наблюдение в динамике до 20 нед гестации.
Затылочное цефалоцеле встречается в 60-80%. В связи с тем, что цефалоцеле окружено мембраной, показатели уровня АФП в крови матери или в амниотической жидкости могут быть нормальными. Постаксиальная полидактилия встречается в 55-75%. Также могут присутствовать другие аномалии конечностей, в виде их искривления и укорочения. Выявление, по крайней мере, двух из трех признаков классической триады при наличии нормального кариотипа позволяет установить диагноз.
Дифференциальный диагноз. Дифференциальный диагноз будет зависеть от типа сочетанных аномалий. Вследствие сходства нескольких эхографических признаков трисомию 13 следует исключить путем кариотипирования. Другим возможным заболеванием является аутосомно-доминантная поликистозная болезнь почек.
Сочетанные аномалии. Перечень возможных аномалий, сочетающихся с данным синдромом, является обширным. В некоторых ситуациях такая широкая фенотипическая вариабельность значительно затрудняет установление диагноза.
Прогноз. Синдром Меккеля (Meckel) является летальным заболеванием. В большинстве случаев отмечаются мертворождения или ранняя неонатальная гибель в течение нескольких часов или дней после рождения. В некоторых наблюдениях описано выживание в течение нескольких месяцев, но с низким качеством жизни. Н.М. Ramadani и Н.А. Nasrat сообщили о ребенке, который умер в возрасте 28 месяцев. В 1997 году P. Paavola et al. привели наблюдение еще одного нетипичного случая выживания в течение 18 месяцев.
Акушерская тактика. При подозрении на синдром Меккеля (Meckel) необходимо провести кариотипирование с целью исключения хромосомных заболеваний. Если принимается решение о ее пролонгировании или если диагноз установлен в более поздние сроки, стандартная акушерская тактика ведения беременной не изменяется.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Новый эхографический признак для изучения нижней челюсти плода в норме и при патологии (микрогнатии) в I триместре беременности (11-14 недель)
Московский областной НИИ акушерства и гинекологии, Москва.
Кафедра медицинской генетики, курс пренатальной диагностики РМАПО, Москва.
Clinica lаs Condes, Santiago, Chili.
Важнейшим маркером генетических синдромов как хромосомного, так и нехромосомного генеза, является микрогнатия. Микрогнатия (нижняя микрогнатия, микрогения) - аномалия развития нижней челюсти, характеризующаяся ее гипоплазией. Диагностика этого состояния при трисомии 18 и триплоидии доходит до 80% [1, 2]. При введении в поисковую систему OMIM термина "micrognatia" можно встретить 447 различных синдромов и ассоциаций, в синдромальное ядро которых входит этот важный генетический маркер. Одна из самых крупных работ в мире по изучению этого маркера принадлежит D. Paladini и соавт. [3], которые описали более 50 случаев микрогнатии в сочетании как с хромосомными [4], так и нехромосомными синдромами и ассоциациями. Степени микрогнатии рассматривались от крайней - агнатии, входящей в состав аутосомно-рецессивного синдрома агнатии, голопрозэнцефалии (отоцефалии) [5, 6]. Отоцефалия - чрезвычайно редкая аномалия, при которой встречаются грубые лицевые дизморфии: недоразвитие или тяжелая гипоплазия нижней челюсти, неправильное положение ушей (рис. 1, 2), которые могут быть объединены и чаще всего располагаются на шее плода [7]. Также крайне выраженная степень микрогнатии может встречаться при окуло-ауриколофронтоназальном синдроме. Он был выделен в самостоятельную нозологическую группу, объединяющую симптомы как фронтоназальной дисплазии, так и синдрома Гольденхара [8, 9].
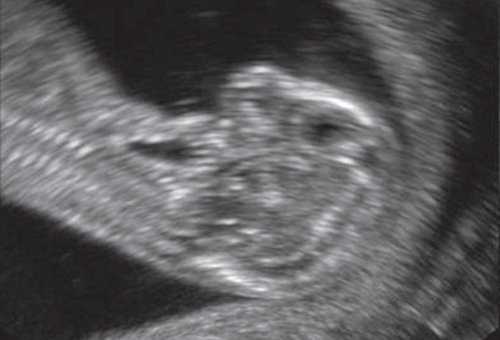
Рис. 1. Профиль плода с синдромом агнатии-голопрозэнцефалии в 12 нед беременности.
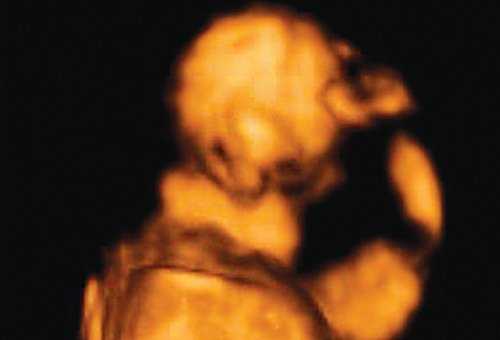
Рис. 2. Фенотип лица плода при синдроме агнатииголопрозэнцефалии в 12 нед беременности.
Гипоплазия нижней челюсти при различных нехромосомных синдромах обычно встречается в сочетании со скелетными дисплазиями и мышечно-скелетными аномалиями: синдром Пьера Робена (рис. 3), Тичера - Коллинза (Франческетти), акрофасциальный дизостоз, цереброкостомандибулярный синдром, ахондрогенез (рис. 4), ателостеогенез, кампомелическая дисплазия, диастрофическая дисплазия (рис. 5), синдром множественных птеригиумов, синдром Пены - Шокейра и др. Наличие микрогнатии характерно для синдрома Карпентера, синдрома Фринса, синдрома Меккеля - Грубера, гидролетального синдрома, синдрома Миллера - Дикера, синдрома Нунан, синдрома Секкеля, Рубинштейна - Тейби и др. Большинство из описанных синдромов имеют аутосомно-рецессивный либо аутосомно-доминантный тип наследования [10, 11].
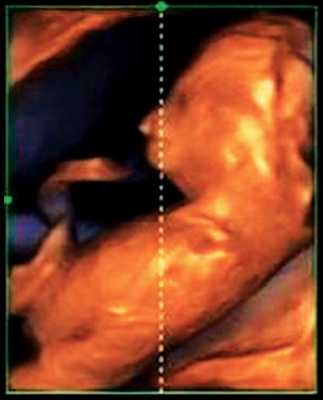
Рис. 3. Микрогнатия у плода при синдроме Пьера Робена в 13 нед беременности.
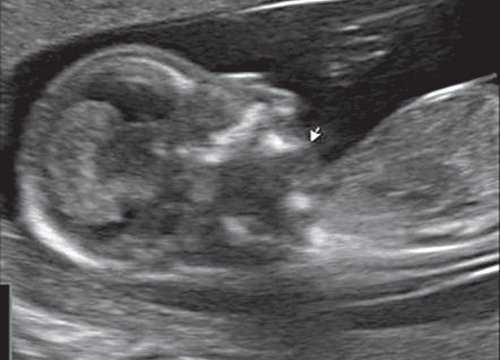
Рис. 4. Микрогнатия у плода с ахондрогенезом в 13 нед беременности.
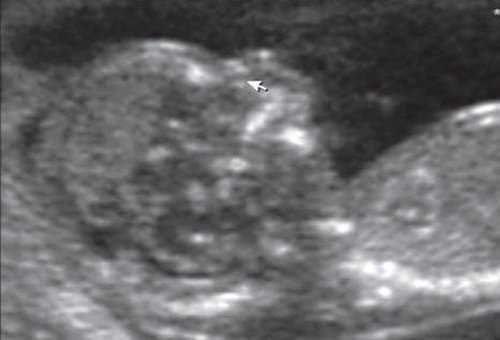
Рис. 5. Микрогнатия у плода с синромом диастрофической дисплазии в 12 нед беременности.
Пренатальная оценка положения и размера нижней челюсти может быть как субъективной, так и объективной. Так, на сегодняшний день известны оценки разных индексов измерения нижней челюсти [3, 6, 12], угла нижней челюсти [13, 14]. Эти измерения сопряжены со значительными погрешностями и в клинической практике применяются не часто, ввиду отсутствия стандартизации изучаемых срезов, трудоемкости и затратности обследования. Учитывая огромную значимость этого маркера, как для диагностики хромосомных, так и нехромосомных синдромов и ассоциаций, поиск новых объективных критериев микрогнатии продолжается 17.
Для качественной оценки особенностей строения нижней челюсти в I триместре беременности специалистами МГО МОНИИАГ совместно с профессором W. Sepulveda (Чили) был изучен и впервые описан новый ультразвуковой признак нижнечелюстной промежуток (mandibular "gap"), визуализируемый при первом скрининговом ("генетическом") ультразвуковом исследовании [18, 19].
Методика базируется на изучении коронарного скана лица плода, так называемого ретроназального треугольника, при котором визуализируется верхняя и нижняя челюсть. Техника получения этого скана чрезвычайно проста и может быть рекомендована для скринингового исследования в 11-14 нед беременности. Эта методика позволяет оценить нижнюю челюсть плода без применения трудоемких оценок, и не сопряжена с математически сложными расчетами коэффициентов, также она существенно не увеличивает время осмотра. Коронарный скан можно оценивать как в режиме 2D, так и в режиме объемной эховизуализации 3D. Методика оценки коронарного скана лица плода в I триместре беременности показана на рисунке 6.
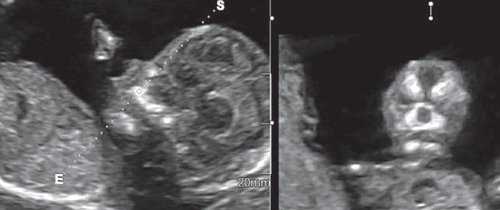
Рис. 6. Методика оценки коронарного скана лица плода в I триместре беременности.
Обе ветви нижней челюсти при сроке 11-14 нед беременности выглядят гиперэхогенными, а в месте слияния имеют характерный гипоэхогенный промежуток, ультразвуковой "разрыв". Этот признак визуализируется при нормальном развитии нижней челюсти (mandibular "gap").
Такие особенности ультразвуковой анатомии связаны с этапами эмбрионального развития костей нижней челюсти, ветви которой начинают развиваться из первой жаберной дуги с 7-й недели эмбрионального развития (рис. 7), и, постепенно приближаясь друг к другу к концу I триместра (на 14-й неделе беременности), образуют синостоз в области подбородка.
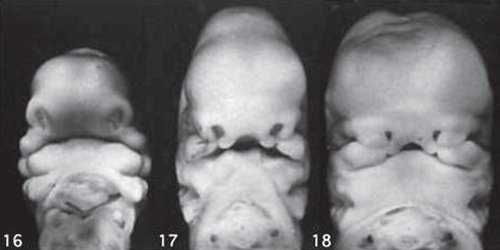
Размер этого промежутка уменьшается с увеличением срока беременности. Нижнечелюстной промежуток здорового плода представлен на рисунке 8.
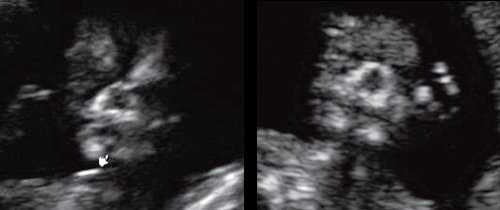
Рис. 8. Нижнечелюстной промежуток в 13 нед беременности при нормальном развитии нижней челюсти.
При патологии нижней челюсти (микрогнатии) в срок 11-14 нед беременности при изучении коронарного скана лица нижнечелюстной "промежуток" отсутствует, нижняя челюсть представлена единой, слившейся костной массой. Отсутствие нижнечелюстного "промежутка" (mandibular "gap") при эхографии в этот срок является маркером гипоплазии нижней челюсти (микрогнатии). Варианты отсутствия нижнечелюстного промежутка при микрогнатии при различных синдромах в срок 11-14 нед беременности представлены на рисунке 9.
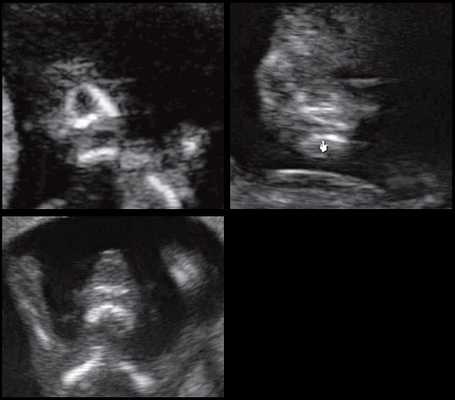
Рис. 9. Отсутствие нижнечелюстного промежутка при микрогнатии, при различных синдромах нехромосомного генеза в 11-14 нед беременности.
Оценка описанного признака при проведении пренатальной эхографии I триместра беременности не только проста в методологии и не требует больших затрат по времени исследования, но и высоко информативна, диагностически точна и специфична.
Литература
- Bianchi D.W., Crombleholme T.M., D'Alton M.E. Micrognathia. In Fetology: Diagnosis and Management of the Fetal Patient // McGraw-Hill: New York. 2000. P. 233-238.
- Nicolaides K.H., Salvesen D.R., Snijders R.J.M., Gosden C. Micrognathia fetal facial defects: Associated malformations and chromosomal abnormalities // Fetal Diagn Ther. 1993. V. 8. Р. 1-9.
- Paladini D., Morra T., Teodoro A., Lamberti A., Tremolaterra F., Martinelli P. Objective diagnosis of micrognathia in the fetus: the Jaw Index // Obstet Gynecol. 1999. V. 93. Р. 382-386.
- Dixon A.D., Hoyte D., Rоnning O. Prenatal development of the facial skeleton // In Fundamentals of Craniofacial Growth. CRC Press: Boca Raton. New York. 1997. Р. 59-97.
- Blaas H.G.K., Eriksson A.G., Salvesen K.A. et al. Brains and faces in holoprosencephaly: pre- and postnatal description of 30 cases // Ultrasound Obstet. Gynecol. 2002. V. 19. 1. P. 24-38.
- Paladini D. Fetal micrognathia: almost always anominous finding // Ultrasound Obstet. Gynecol. 2010. V. 35. P. 377-384.
- Cohen M.M.Jr. Perspectives on holoprosencephaly: Part I. Epidemiology, genetics, and syndromology // Teratology. 1989. V. 40. Р. 211-235.
- Carey J.C., Yong S.L. Frontonasal dysplasia and Goldenhar syndrome: the oculo-auriculo-frontonasal syndrome // Paper presented at the Conference on Malformations and Morphogenesis (March of Dimes). Dartmouth College, Hanover, NH, USA. 1981.
- Casey H.D., Braddock S.R., Haskins R.C., Carey J.C., Morales L. Frontonasal malformation and the oculoauriculovertebral spectrum: the oculoauriculofrontonasal syndrome // Cleft Palate Craniofac. J. 1996. V. 33. Р. 519-523.
- Ван Фехт Дж. Ультразвуковые маркеры хромосомных аномалий у плода // Ультразвуковая Диагностика. 1997. 3. С. 37-44.
- Turner G.M., Twining P. The facial profile in the diagnosis of fetal abnormalities // Clin Radiol. 1993. V. 47. Р. 389-395.
- Chitty L.S., Campbell S., Altman D.G. Measurements of the fetal mandible feasibility and construction of a centile chart // Prenat Diagn. 1993. V. 13. Р. 749-756.
- Otto C., Platt L.D. The fetal mandible measurement: an objective determination of fetal jaw size // Ultrasound Obstet Gynecol. 1991. V. 1. Р. 12-17.
- Rotten D., Levaillant J.M., Martinez H., Ducou H., Le Pointe D., Vicaut E. The fetal mandible: a 2D and 3D sonographic approach to the diagnosis of retrognathia and micrognathia // Ultrasound Obstet. Gynecol. 2002. V. 19. Р. 122-130.
- Bronshtein M., Blazer S., Zalel Y., Zimmer E.Z. Ultrasonographic diagnosis of glossoptosis in fetuses with Pierre Robin sequence in early and mid pregnancy // Am. J. Obstet. Gynecol. 2005. V. 193. Р. 1561-1564.
- Chitty L.S., Campbell S., Altman D.G. Measurements of the fetal mandible feasibility and construction of a centile chart // Prenat Diagn. 1993. V.13. Р. 749-756.
- Watson W. J., Katz V.L. Sonographic measurement of the fetal mandible: standards for normal pregnancy // Am J Perinatol. 1993. V. 10. Р. 226-228.
- Sepulveda W., Wong A., Andreeva E., Adzehova N. Absent mandibular gap at retronasal triangle view: a clue to the diagnosis of micrognathia in the first trimester // Ultrasound in obstetrics and gynecology. 2012. V. 39. P. 152-156.
- Sepulveda W., Wong A., Andreeva E. et al. A novel, simple technique for diagnosis of micrognathia in firsttrimester: identification of the receding chin on the retronasal triangle (RNT) view. Oral poster abstracts. 21 World Congress on Obstetrics and Gynecology. LosAngeles // Ultrasound in Obstetrics and Gynecology. 2011. V. 38. P. 64.
УЗИ сканер HS60
Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Читайте также:
- Показания, подготовка к хондропластике и микрофрактурингу коленного сустава
- Закупорка центральных вен сетчатки и их ветвей
- Консервативная терапия острой артериальной непроходимости. Неоперативное лечение тромбоза
- Причины снижения кровотока в миокарде. Окклюзивный атеросклероз.
- Септо-оптическая дисплазия - клиника, диагностика