Синдром Оменна. Имммунодефицит при дефиците антигенов МНС
Добавил пользователь Владимир З. Обновлено: 27.01.2026
2. У больных наблюдаются эритродермия, лимфаденопатия, гепатоспленомегалия и упорный понос. В некоторых случаях обнаружены мутации
Синдром Оменна представляет собой аутосомно-рецессивное
фатальный комплеусный иммунодефицит, с резко повышенной
восприимчивостью к инфекциям и инфильтрацией кожи,
кишечника, печени и селезенки Т-лимфоцитами.
У больных наблюдаются эритродермия,
лимфаденопатия, гепатоспленомегалия и
упорный понос. В некоторых случаях
обнаружены мутации генов, кодирующих
RAG1 и RAG2.
RAG гены — группа генов, детерминирующих структуру и биосинтез
рекомбиназ. Эти гены, находятся на хромосоме 11р13, участвуют в
процессах соматической перестройки генов TCR (т-клеточный
рецептор) и BCR( В-клеточный рецептор).
Кодируют два белка — RAG- 1и RAG-2, участвующих в перестройках
генетических сегментов, кодирующих рецепторы иммунных клеток.
И регулирующие V(D)J-рекомбинации.
Есть две формы:
o Полный дефицит RAG1/RAG2 встречается редко и приводит к
нарушению развитию отсутствию злых форм Т и В лимфоцитов,
o Мисенс мутация RAG1/RAG2 возникает не полный
иммунодефицит. И функция RAG1/RAG2 частично сохранена.
4. В процессе V(D)J-перестройки генные сегменты, по одному из каждого класса, соединяются вместе. Объединенная последовательность
V(D)J-рекомбинация — механизм соматической рекомбинации ДНК,
происходящий на ранних этапах дифференцировки лимфоцитов и приводящий к
формированию антиген-распознающих участков иммуноглобулинов и Тклеточного рецептора.
Гены иммуноглобулина и Т-клеточного рецептора состоят из повторяющихся
сегментов, принадлежащих к трем классам: V (variable), D (diversity) и J (joining).
В процессе V(D)J-перестройки
генные сегменты, по одному из
каждого класса, соединяются
вместе. Объединенная
последовательность сегментов
V(D)J кодирует вариабельные
домены каждой из
цепей рецептора или
иммуноглобулина.
хромосома
11р13
Синтез на РНК
белка-рекомбиназы
RAG1/RAG2
пpe-BCR и пpe-TCR
V(D)J-перестройки
Пре-Вклетки
Зрелые
В-клетки
V(D)J-перестройки
Пре-Тклетки
Зрелые
Т-клетки
У больных синдромом Омена грудного возраста находят
постоянный лейкоцитоз с выраженной эозинофилией и
лимфоцитозом, повышенный уровень IgE, низкое
содержание IgG, IgA и IgM в сыворотке, почти полное
отсутствие. В-лимфоцитов и увеличенное число
аутореактивных Т-лимфоцитов. Преобладают Тh2-клетки
с резко нарушенной функции.
В крови больных присутствует очень мало CD4 Т-лимфоцитов, но число CD8
Т-лимфоцитов нормально или даже повышено. Лимфопения выражена
лишь умеренно.
На В-лимфоцитах (несмотря на нормальное их число) и моноцитах не
удается обнаружить антигенов МНС класса II (HLA-DP, -DQ и -DR). Отсутствие
этих антигенпредставляющих молекул нарушает реакцию на различные
антигены, и поэтому у больных отмечается гипогаммаглобулинемия.
Кроме того, В-лимфоциты, лишенные антигенов МНС класса II, не
стимулируют аллогенные клетки в смешанной культуре лейкоцитов.
Пролиферативный ответ лимфоцитов на митогены сохраняется, но ответ на
антигены отсутствует.
Характерна резкая гипоплазия тимуса и других лимфоидных органов.
Отсутствие молекул МНС класса II нарушает процессы селекции в тимусе и
изменяет экспрессию комплекса TCR-CD3 на CD4 Т-лимфоцитах.
8. Клинические проявления
•Генерализованная эритродермия
•Гепато-cпленомeгалия
•Лихорадка
• Хроническая диарея
•Плохая прибавка массы тела
•Частая заболеваемостьэнтеровирусными инфекциями,
кандидоз и др.
•аллопеции
При по Полном дефиците
RAG1/RAG2 (ТКИД)
oПри вакцинации БЦЖдиссеминация инфекции.
oПри переливание не облученной
крови развитие реакции
«трансплантат против хозяина».
При по частичном дефиците
RAG1/RAG2
o Развитие диссеминации после
прививки БЦЖ не происходит
o Не развивается реакции
«трансплантат против хозяина».
o Развитие генерализованного
отека.
o Выраженная воспалительная
инфильтрация приводит к
развитию склеродермии.
10. Лабораторные исследования
В общем анализе крови
•анемия
• тромбоцитопения
•лейкоцитоз с выраженной эозинофилией (конмпенсаторной) и
лимфоцитозом (за счет Th2 –клеток).
В биохимический анализ крови
•различные уровни активированных циркулирующих Т- клеток с очень
низким ответом на стимуляцию антигенами и различным ответом на ФГА.
•отсутствие или очень низкий уровень В-клеток.
•Определяется гипопротеинемия, гипогаммаглабулинемия (А. М. G), при
повышенном уровне общего IgE, Уровень сывороточных IgA, IgM, IgG
значительно снижен.
12. Специальные исследования
•На рентгенограме Тимус гипоплазирован, со значительным
истощением лимфоидного компонента и часто отсутствующими
тельцами Гассаля.
•При биопсии кожи обнаруживают лимфоцитарные инфильтраты,
иногда обнаруживают гистиоциты и эозинофилы. Лимфоцитарные
инфильтраты состоят активированных Т-клеток (CD45RO, DR),
некоторые также экспрессируют СD 30.
•Увеличенных лимфатических узлах отсутствуют фолликулы, истощена
популяция нормальных лимфоцитов и увеличено количество
эoзинофилов и ретикулярных клеток.
13. Лечение
Для лечения используют различные иммуносупрессивные препараты.
Неплохой эффект показало применение циклоспорина А,
Применение стероидов малоэффетивно,
Показан положительный эффект от ежедневного применения ИНФ-y одного
ребенка, что сопровождалось клиническим улучшением, снижением
эозинофилов и усиление пролиферативного ответа лимфоцитов на митогены.
Синдром Оменна. Имммунодефицит при дефиците антигенов МНС
Синдром Оменна. Имммунодефицит при дефиците антигенов МНС
Синдром Оменна представляет собой аутосомно-рецессивное фатальное состояние с резко повышенной восприимчивостью к инфекциям и инфильтрацией кожи, кишечника, печени и селезенки Т-лимфоцитами. У больных наблюдаются эритродермия, лимфаденопатия, гепатоспленомегалия и упорный понос. В некоторых случаях обнаружены мутации генов, кодирующих RAG1 и RAG2. Таким образом, этот синдром является одной из форм тяжелого комплексного иммунодефицита.
У больных синдромом Омена грудного возраста находят постоянный лейкоцитоз с выраженной эозинофилией и лимфоцитозом, повышенный уровень IgE, низкое содержание IgG, IgA и IgM в сыворотке, почти полное отсутствие В-лимфоцитов и увеличенное число аутореактивных Т-лимфоцитов. Преобладают Тh2-клетки с резко нарушенной функцией.
Иммунодефицит при дефиците антигенов МНС класса I
Нарушение экспрессии антигенов главного комплекса гистосовместимости включают нарушение экспрессии антигенов МНС классов I (HLA-A, -В и -С) и II (HLA-DR, -DQ и -DP). Связь гуморального и клеточного иммунодефицита с недостаточностью HLA подчеркивает важнейшее значение этих антигенов для эффективной кооперации клеток иммунной системы.
Изолированный дефицит антигенов МНС класса I, или синдром обнаженных лимфоцитов, встречается редко. Нарушения иммунитета при этом выражены в гораздо меньшей степени, чем при ТКИД, и проявляются в более позднем возрасте. В сыворотке крови больных детей содержится нормальное количество антигенов МНС класса I и b2-микроглобулина, но на всех клетках эти антигены отсутствуют. Отмечается дефицит CD8, но не CD4 Т-лимфоцитов. Обнаружены мутации двух генов в локусе МНС на хромосоме 6, которые кодируют транспортеры пептидов ТАР1 и ТАР2.
ТАР переносят антигенные пептиды из цитоплазмы через мембраны аппарата Гольджи, и присоединяют их к а-цепи антигенов МНС класса I и b2-микроглобулину. Все эти компоненты собираются в комплекс МНС класса 1, который затем перемещается на поверхность клеток. Незавершенный в отсутствие антигенного пептида комплекс разрушается в цитоплазме.
Иммунодефицит при нехватке антигенов МНС класса II
Многие больные с дефицитом антигенов МНС класса II (HLA-DR, -DQ и -DP) принадлежат к потомкам североафриканцев. В раннем грудном возрасте развивается упорный понос, часто связанный с криптоспоридиозом и энтеровирусными инфекциями (вирусы полиомиелита и Коксаки). Повышена частота и других вирусных инфекций, кандидозного стоматита, бактериальной пневмонии и септицемии. Нарушения иммунитета выражены слабее, чем при ТКИД. Так, вакцинация БЦЖ не приводит к диссеминации инфекции, а при переливании необлученной крови не происходит реакции «трансплантат против хозяина».
Установлены четыре разных молекулярных дефекта, приводящих к нарушению экспрессии антигенов МНС класса II. В основе одной из форм этой патологии лежит мутация гена, расположенного на длинном плече хромосомы 1 (lq) и кодирующего белок RFX5. RFX5 представляет собой субъединицу фактора транскрипции RFX, взаимодействующего с Х-боксом промоторов генов МНС класса П. Вторая форма обусловлена мутацией гена, расположенного на длинном плече хромосомы 13 (13q) и кодирующего вторую субъединицу комплекса RFX — RFX-ассоциированный белок (RFXAP) с молекулярной массой 36 кДа. Позже обнаруженной и самой частой причиной дефицита антигенов МНС класса II являются мутации гена RFXANK, кодирующего третью субъединицу того же комплекса.
При четвертой форме выявлена мутация расположенного на хромосоме 16 (участок р13) гена, который кодирует трансактиватор СИТА генов МНС класса II. Этот не связывающийся с ДНК коактиватор контролирует клеточную специфичность и индукцию антигенов МНС класса II. Все эти молекулярные дефекты нарушают координированную экспрессию антигенов МНС класса II на поверхности В-лимфоцитов и макрофагов.
В крови больных присутствует очень мало CD4 Т-лимфоцитов, но число CD8 Т-лимфоцитов нормально или даже повышено. Лимфопения выражена лишь умеренно. На В-лимфоцитах (несмотря на нормальное их число) и моноцитах не удается обнаружить антигенов МНС класса II (HLA-DP, -DQ и -DR). Отсутствие этих антигенпредставляющих молекул нарушает реакцию на различные антигены, и поэтому у больных отмечается гипогаммаглобулинемия. Кроме того, В-лимфоциты, лишенные антигенов МНС класса II, не стимулируют аллогенные клетки в смешанной культуре лейкоцитов.
Пролиферативный ответ лимфоцитов на митогены сохраняется, но ответ на антигены отсутствует. Характерна резкая гипоплазия тимуса и других лимфоидных органов. Отсутствие молекул МНС класса II нарушает процессы селекции в тимусе и изменяет экспрессию комплекса TCR-CD3 на CD4 Т-лимфоцитах.
Синдром Оменна. Имммунодефицит при дефиците антигенов МНС
Специфические и расширенные лабораторные тесты при иммунодефиците*
Определение уровня IgE
У пациентов с неполным дефектом или дефицитами В-клеток уровни могут быть высокими или низкими.
Изолированный дефицит не имеет значимых клинических проявлений.
Определение количества В-клеток методом проточной цитометрии
Низкие уровни Ig
Отсутствие В-клеток при синдроме Оменна.
Для некоторых пациентов с лимфаденопатией, для определения, являются ли зародышевые центры нормальными, и исключения наличия онкозаболеваний и инфекций
Интерпретация зависит от результатов гистологических исследований.
Генетическое тестирование (генетическое секвенирование или поиск мутаций)†
Количество В-клеток 1% (определяется методом проточной цитометрии)
Подозрение нарушения с одной или несколькими характерными мутациями
Наличие дефектов в генах предполагает или подтверждает следующие диагнозы, как в следующих нескольких из многих примеров:
BTK: Х-сцепленная агаммаглобулинемия
NEMO: Комбинированный иммунодефицит
Результаты могут также использоваться для прогностической информации.
Определение количества Т-клеток методом проточной цитометрии и моноклональных антител§
Интерпретация зависит от молекулярной основы данного типа ТКИД.
Анализ пролиферации Т-клеток в ответ на стимуляцию митогенами, антигенов, или облученных аллогенных лейкоцитов
Низкий процент Т-клеток, лимфопения, подозрение на ТКИН или «полный» синдром Ди Джорджи
Низкое или отсутствующее поглощение радиоактивного тимидина в течение клеточного деления указывает на Т-клетки или комбинированный дефект.
Обнаружение антигенов (например, класс II молекул MHC) с использованием моноклональных антител или серологического HLA-типирования
Подозреваемый дефицит МНС, отсутствие стимуляции МНС со стороны клеток
Отсутствие HLA антигенов I или II класса, определенное методом серологического HLA-типирования, является диагностическим показателем дефицита антигена MHC.
Количественный анализ аденозина дезаминазы в эритроцитах
Низкие уровни при специфической форме ТКИД.
Количественное определение пурин нуклеозидфосфорилазы
Тяжелая персистирующая лимфопения
Низкие уровни с комбинированным иммунодефицитом при нормальном или повышенном уровне Ig.
Анализ рецепторов Т-клеток и сигнальной трансдукции
Фенотипически нормальные Т-клетки, которые обычно не пролиферируют в ответ на воздействие митогена и антигена
Интерпретация зависит от вида теста.
Определение Т-рецепторных эксцизионных колец (TREC)
Скрининг на ТКИД и другие нарушения Т-клеток
Низкие значения указывают на дефект, нарушающий развитие или созревание Т-клеток или вызывающий апоптоз Т-клеток.
Комбинированный гуморальный и клеточный иммунодефицит
Подозрение на комбинированный иммунодефицит
Наличие дефектов в генах предполагает или подтверждает определенные нарушения; например, аномалии NEMO предполагают собой комбинированный иммунодефицит с нарушениями регулирования NF-каппа B, а нарушения IL-2RG предполагают ТКИД.
Дефекты фагоцитарной системы
Анализ продукции оксидантов (перекись водорода, супероксид) или белков (CR3 [CD11], адгезивных гликопротеинов, компонентов НАДФН-оксидазы)
Стафилококковые абсцессы или инфекции определенными грамотрицательными бактериями или грибками в анамнезе (например, аспергиллез, инфекции Serratia marcescens)
Аномалии подтвердить дефекты или дефициты фагоцитарных клеток.
Анализы фосфорилирования переносчика сигнала и активатора транскрипции (STAT), в том числе STAT1 и STAT4
Рецидивирующие микобактериальные инфекции
Этот тест проводят первым для проверки наличия Менделевской предрасположенности к микобактериальным инфекциям (МПМИ).
Исследование специфики уровней компонентов комплемента
Подозрение на дефицит комплемента
Интерпретация зависит от вида теста.
*Некоторые из этих тестов могут использоваться при скрининге или предварительном тестировании.
† Коммерчески доступными являются генетические панели для выявления первичных иммунодефицитов и специфических заболеваний, таких как общий вариабельный иммунодефицит (CVID) или тяжелый комбинированный иммунодефицит (ТКИД). For information on interpreting genetic studies in these disorders, see Chinn IK, Chan AY, Chen K, et al: Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol 145(1):46–69, 2020. doi: 10.1016/j.jaci.2019.09.009
‡ SAP, также называемый доменом SH2 белка 1А [SH2D1A] или DSHP.
§В тесте используется анти-CD3 для всех Т-клеток, анти-CD4 для Т-хелперов, анти-CD8 для цитотоксических Т-клеток, анти-CD45RO или анти-CD45RA для активированных и интактных Т-клеток, анти-CD25 для регуляторных Т-клеток и анти-CD16 и анти-CD56 для естественных клеток-киллеров.
ВТК = тирозинкиназа Брутона; СН = гемолитический комплемент;РК (CR) = рецептор комплемента; ВНКИ (CVID) = вариабельный неклассифицируемый иммунодефицит; ГКГС (HLA) = главный комплекс гистосовместимости человека; Ig = иммуноглобулин; IL2RG = интерлейкин-2 гамма-рецептор; МНС = главный комплекс гистосовместимости; НАДФН = никотинамидадениндинуклеотидфосфат; NEMO = NF-каппа-В незаменимый модулятор; RBC = эритроцит; NF-каппа-B = ядерный фактор каппа-B; SAP = SLAM-ассоциированный белок; SCID = тяжелый комбинированный иммунодефицит; SLAM = сигнальная лимфоцит-активирующая молекула, WBC = лейкоцит.
Презентация на тему Синдром Омена
Слайды и текст этой презентации

Студент: Юровских К. С.
339 группа
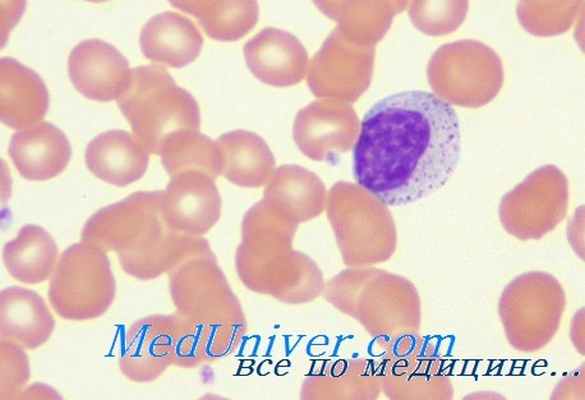
У больных наблюдаются эритродермия, лимфаденопатия, гепатоспленомегалия и упорный понос. В некоторых случаях обнаружены мутации генов, кодирующих RAG1 и RAG2.
Синдром Оменна представляет собой аутосомно-рецессивное фатальный комплеусный иммунодефицит, с резко повышенной восприимчивостью к инфекциям и инфильтрацией кожи, кишечника, печени и селезенки Т-лимфоцитами.

RAG гены — группа генов, детерминирующих структуру и биосинтез рекомбиназ. Эти гены, находятся на хромосоме 11р13, участвуют в процессах соматической перестройки генов TCR (т-клеточный рецептор) и BCR( В-клеточный рецептор).
Кодируют два белка — RAG- 1и RAG-2, участвующих в перестройках генетических сегментов, кодирующих рецепторы иммунных клеток. И регулирующие V(D)J-рекомбинации.
Есть две формы:
Полный дефицит RAG1/RAG2 встречается редко и приводит к нарушению развитию отсутствию злых форм Т и В лимфоцитов,
Мисенс мутация RAG1/RAG2 возникает не полный иммунодефицит. И функция RAG1/RAG2 частично сохранена.
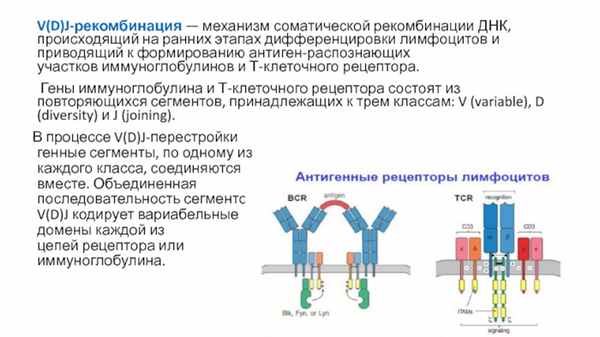
В процессе V(D)J-перестройки генные сегменты, по одному из каждого класса, соединяются вместе. Объединенная последовательность сегментов V(D)J кодирует вариабельные домены каждой из цепей рецептора или иммуноглобулина.
V(D)J-рекомбинация — механизм соматической рекомбинации ДНК, происходящий на ранних этапах дифференцировки лимфоцитов и приводящий к формированию антиген-распознающих участков иммуноглобулинов и Т-клеточного рецептора.
Гены иммуноглобулина и Т-клеточного рецептора состоят из повторяющихся сегментов, принадлежащих к трем классам: V (variable), D (diversity) и J (joining).
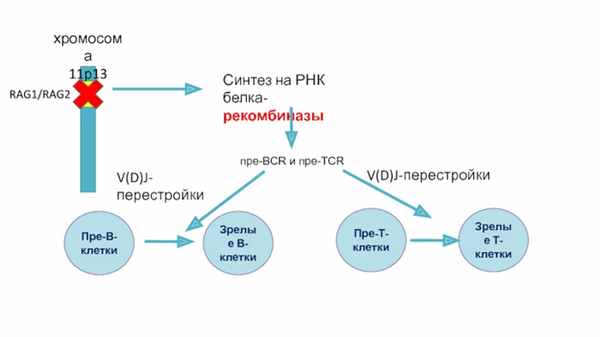
Синтез на РНК
белка-рекомбиназы
пpe-BCR и пpe-TCR

У больных синдромом Омена грудного возраста находят постоянный лейкоцитоз с выраженной эозинофилией и лимфоцитозом, повышенный уровень IgE, низкое содержание IgG, IgA и IgM в сыворотке, почти полное отсутствие. В-лимфоцитов и увеличенное число аутореактивных Т-лимфоцитов. Преобладают Тh2-клетки с резко нарушенной функции.

В крови больных присутствует очень мало CD4 Т-лимфоцитов, но число CD8 Т-лимфоцитов нормально или даже повышено. Лимфопения выражена лишь умеренно.
На В-лимфоцитах (несмотря на нормальное их число) и моноцитах не удается обнаружить антигенов МНС класса II (HLA-DP, -DQ и -DR). Отсутствие этих антигенпредставляющих молекул нарушает реакцию на различные антигены, и поэтому у больных отмечается гипогаммаглобулинемия.
Кроме того, В-лимфоциты, лишенные антигенов МНС класса II, не стимулируют аллогенные клетки в смешанной культуре лейкоцитов. Пролиферативный ответ лимфоцитов на митогены сохраняется, но ответ на антигены отсутствует.
Характерна резкая гипоплазия тимуса и других лимфоидных органов. Отсутствие молекул МНС класса II нарушает процессы селекции в тимусе и изменяет экспрессию комплекса TCR-CD3 на CD4 Т-лимфоцитах.

Генерализованная эритродермия
Гепато-cпленомeгалия
Лихорадка
Хроническая диарея
Плохая прибавка массы тела
Частая заболеваемость- энтеровирусными инфекциями, кандидоз и др.
аллопеции
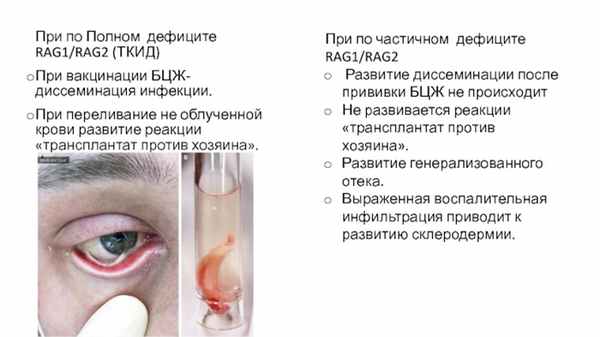
При по Полном дефиците RAG1/RAG2 (ТКИД)
При вакцинации БЦЖ- диссеминация инфекции.
При переливание не облученной крови развитие реакции «трансплантат против хозяина».
При по частичном дефиците RAG1/RAG2
Развитие диссеминации после прививки БЦЖ не происходит
Не развивается реакции «трансплантат против хозяина».
Развитие генерализованного отека.
Выраженная воспалительная инфильтрация приводит к развитию склеродермии.

В общем анализе крови
анемия
тромбоцитопения
лейкоцитоз с выраженной эозинофилией (конмпенсаторной) и лимфоцитозом (за счет Th2 –клеток).

В биохимический анализ крови
различные уровни активированных циркулирующих Т- клеток с очень низким ответом на стимуляцию антигенами и различным ответом на ФГА.
отсутствие или очень низкий уровень В-клеток.
Определяется гипопротеинемия, гипогаммаглабулинемия (А. М. G), при повышенном уровне общего IgE, Уровень сывороточных IgA, IgM, IgG значительно снижен.

На рентгенограме Тимус гипоплазирован, со значительным истощением лимфоидного компонента и часто отсутствующими тельцами Гассаля.
При биопсии кожи обнаруживают лимфоцитарные инфильтраты, иногда обнаруживают гистиоциты и эозинофилы. Лимфоцитарные инфильтраты состоят активированных Т-клеток (CD45RO, DR), некоторые также экспрессируют СD 30.
Увеличенных лимфатических узлах отсутствуют фолликулы, истощена популяция нормальных лимфоцитов и увеличено количество эoзинофилов и ретикулярных клеток.

Для лечения используют различные иммуносупрессивные препараты.
Неплохой эффект показало применение циклоспорина А,
Применение стероидов малоэффетивно,
Показан положительный эффект от ежедневного применения ИНФ-y одного ребенка, что сопровождалось клиническим улучшением, снижением эозинофилов и усиление пролиферативного ответа лимфоцитов на митогены.
Тяжелый первичный комбинированный иммунодефицит: трудности диагностики и фатальность исхода
Е.Н. Охотникова1, Ю.И. Гладуш2, Т.П. Иванова2, Е.И. Усова1, Е.В. Поночевная1, О.Ф. Зарудняя2, В.Т. Леуш2, С.А. Онысько2, Е.В. Макаревская2 1Национальная медицинская академия последипломного образования имени П.Л. Шупика, 2Национальная детская специализированная больница «ОХМАТДЕТ»
В настоящее время общеизвестно, что первичные иммунодефицитные состояния (ПИДС) – не столь редкая патология, как было принято считать еще 20-30 лет назад. Однако, несмотря на совершенствование методов диагностики иммунодефицитных состояний (ИДС), более чем у 70% больных выявить их не удается [5, 6]. Поэтому диагностика ПИДС и идентификация их нозологических форм, определение иммунного статуса больных, в первую очередь с точки зрения обеспечения адекватной терапии, стали актуальными проблемами клинической медицины и особенно педиатрии [2, 3].
| ПИДС – это наследственно обусловленные заболевания, вызванные нарушением сложного каскада реакций, необходимых для удаления чужеродных агентов из организма и развития адекватных воспалительных реакций [1]. Их типичными проявлениями являются тяжело протекающие бактериальные, вирусные и грибковые инфекции, аутоиммунные заболевания и повышенная склонность к развитию злокачественных новообразований. |
В настоящее время описано более 80 форм ПИДС. Частота встречаемости ПИДС составляет от 1 на 1 тыс. до 1 на 5 млн населения в зависимости от формы.
Диагноз ПИДС можно установить только после специального обследования с использованием иммунологических, генетических и молекулярных методов исследования. Это важно сделать как можно раньше, чтобы предотвратить тяжелые необратимые последствия ПИДС.
Инфекционный синдром ИДС, как правило, является ведущим в клинической картине этой патологии, однако предрасположенность к инфекциям – это один из основных, но далеко не единственный, а в ряде случаев даже второстепенный и не первый по времени возникновения симптомокомплекс.
Неинфекционные клинические проявления, часто встречающиеся при ИДС и достаточно подозрительные с точки зрения диагностики, особенно если они сочетаются с инфекциями, могут быть следующими: диарея и значительное отставание в физическом развитии, артропатии и синдром, подобный системной красной волчанке, гепатоспленомегалия и гематологические изменения (например агранулоцитоз), аплазия или гиперплазия лимфоидной ткани (неинфекционная и неонкологическая лимфаденопатия, спленомегалия, гипергаммаглобулинемия), идиопатические эндокринопатии (в том числе гипопаратиреоз) и неврологические нарушения (ментальная недостаточность, микроцефалия, атаксия), признаки преждевременного старения (например седые волосы) и склонность к онкологическим заболеваниям (у больного и у членов его семьи), скелетные дефекты и пороки развития сердца. Все эти симптомы могут встречаться при многих ИДС [3].
Клинические и иммунологические особенности некоторых нозологических форм ИДС представлены ниже [3, 4].
Тяжелый комбинированный иммунный дефицит (SCID)
Наиболее серьезным заболеванием из группы комбинированных дефектов иммунитета является тяжелый комбинированный иммунный дефицит (ТКИД), который проявляется в первые месяцы жизни. ТКИД представляет собой группу генетически разнородных заболеваний, в основе которых лежит нарушение созревания Т-лимфоцитов с полным отсутствием их функции. В зависимости от генетического дефекта заболевание наследуется Х-сцепленно или аутосомно-рецессивно.
| Критериями диагноза для всех форм ТКИД являются гипоплазия лимфоидной ткани, лимфопения, выраженное снижение уровня CD3+-клеток, значительное уменьшение концентраций сывороточных иммуноглобулинов, раннее начало тяжелых инфекций. |
В зависимости от формы заболевания количество В-лимфоцитов варьирует от нулевых (Т-В-) до нормальных значений (Т-В+), однако во всех случаях их функция значительно нарушена. При некоторых формах ТКИД определяется нормальное количество натуральных киллерных клеток (НК+).
К типичным клиническим проявлениям ТКИД относятся задержка физического развития, хроническая диарея, тяжелая молочница и грибковые поражения кожи, прогрессирующее поражение респираторного тракта в виде пневмоциcтных пневмоний, вирусных и бактериальных инфекций, сепсиса и пневмонии.
ТКИД иммунологически характеризуются нарушением дифференцировки стволовых клеток, блоком созревания Т- и В-лимфоцитов и их дефицитом. Комбинированные формы иммунодефицита встречаются чаще, чем селективные. Как правило, они связаны с нарушением функции центральных органов иммунной системы (вилочковой железы и костного мозга). При комбинированных ИДС ведущая роль принадлежит дефекту Т-клеток.
1. Cиндром ретикулярной дисгенезии – характеризуется уменьшением количества стволовых клеток в костном мозге. Для указанного синдрома характерна внутриутробная гибель плода или смерть детей вскоре после рождения.
2. «Швейцарский» тип иммунодефицита – характеризуется поражением Т- и В-систем и, следовательно, нарушением клеточных и гуморальных реакций иммунологической защиты. При аутосомно-рецессивных формах, по крайней мере, у 40% больных обнаруживается недостаточность аденозиндезаминазы, что ведет к нарушению метаболизма аденозина, блокаде синтеза гипоксантина и в результате этого – блокаде созревания Т-клеток. У части детей на мембранах Т-лимфоцитов отсутствуют антигены гистосовместимости (МНС) 1-го или 2-го класса. Содержание В-клеток может соответствовать нормальному уровню или превышать его, но эти клетки не способны секретировать иммуноглобулины (Ig) в достаточном количестве.
Заболевание проявляется в первые месяцы жизни и часто характеризуется злокачественным течением. Наблюдается задержка прибавки массы тела, уже в первые дни жизни у некоторых детей появляются кореподобные высыпания на коже, что может быть связано с реакциями несовместимости по отношению к материнским лимфоцитам, поступающим через плаценту в кровоток ребенка. Развиваются признаки кожного кандидоза, диарея, острая интерстициальная пневмония, приобретающая затяжной и рецидивирующий характер. Такие дети очень восприимчивы к вирусным инфекциям. В крови выявляется значительная лимфопения, особенно низким является содержание Т-лимфоцитов. Уровень Ig всех классов заметно снижен. Исключение составляют грудные дети с IgG, полученными от матери. Патогномоничны изменения вилочковой железы, гипоплазия миндалин и лимфатических узлов. Возникает неспособность проявлять реакции гиперчувствительности замедленного типа. Дети редко доживают до 2-летнего возраста.
Дифференциальная диагностика
Дифференциальную диагностику проводят в направлении исключения или подтверждения следующих заболеваний:
• ВИЧ-инфекции;
• врожденной краснухи;
• ПИДС:
• синдрома Ди Джорджи;
• дефицита ZAP70;
• CD3-дефицита;
• гипоплазии хрящей и волос;
• дефицита молекул HLA II класса;
• дефицита пуриннуклеозид-фосфорилазы [1].
В качестве иллюстрации приводим из собственной практики случай ТКИД у ребенка грудного возраста, демонстрирующий особенности течения заболевания и трудности его дифференциальной диагностики.
| В плане подозрения на ПИДС обращали на себя внимание следующие клинические проявления: 1. Дебют заболевания, которое характеризовалось быстро прогрессирующим злокачественным течением, – в 2,5 мес жизни. 2. Значительный интоксикационный синдром, обусловленный тяжелейшим микст-бактериальным процессом: сепсисом туберкулезной и синегнойной этиологии с развитием полиорганного поражения (БЦЖ-ит, деструкция легочной ткани, энтерит, гепатит). 3. Выраженный гепатолиенальный синдром: печень – до 5,5 см ниже реберной дуги, плотная, край острый; селезенка нижним полюсом лежит на гребне подвздошной кости, плотная, гладкая (что явилось основанием для исключения онкогематологической патологии). 4. Массивный прогрессирующий гематологический синдром: анемия, тромбоцитопения и лейкопения (4,0-2,7-2,2х10 9 /л) с анэозинофилией, нейтропенией и абсолютной лимфопенией (1,386-0,99х10 9 /л) вплоть до панцитопении с прогрессирующим некупируемым ДВС-синдромом (тромбоцитопения, некрозы кожи и подкожной жировой клетчатки, некротический энтероколит, развитие полиорганной недостаточности, сладж-феномен и др.). 5. Развитие вторичного гистиоцитарного синдрома с накоплением во всех органах и тканях миелоидных клеток, что затруднило диагностику основного заболевания. 6. Невыявление патологии иммунных органов (хотя и подозревалась) – вилочковой железы и лимфоидных образований – с помощью УЗИ и КТ, что еще раз свидетельствует об их низкой диагностической ценности. 7. Иммунологический синдром не был типичным, что затруднило прижизненную диагностику ПИДС и не позволило идентифицировать его вариант. |
Таким образом, современная медицина располагает средствами терапии больных с врожденными дефектами иммунной системы, однако в Украине до настоящего времени нет технических и финансовых возможностей для ранней диагностики и адекватного лечения детей с ПИДС, что объясняет крайне низкую их выявляемость и высокую летальность пациентов. Следует отметить, что даже в экономически развитых странах ПИДС выявляют лишь у 30% больных, и они умирают от септических, онкологических, неврологических, аутоиммунных или других заболеваний. Патогенетическая, клиническая и прогностическая вариабельность ПИДС делает их терапию достаточно сложной. К сожалению, в настоящее время выбор тактики и объема лечения, как правило, базируется не столько на оценке состояния больного, сколько на кумулятивном опыте работы ведущих центров клинической иммунологии, накопленных ими данных о влиянии на течение и исход болезни различных методов терапии [7, 8].
Однако наука не стоит на месте, и скорость внедрения новых технологий не позволяет считать безнадежными даже больных с наиболее тяжелыми вариантами ПИДС.
Читайте также: