Синдром Пена-Шокейра. Пентада Кантрелла
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Актульность. Пентада Кантрелла – редкий комбинированный порок развития, представленный пятью признаками: торакоабдоминальным дефектом передней брюшной стенки, дефектами нижней части грудины, передней части диафрагмы и диафрагмального перикарда, а также врожденными аномалиями сердца. Прогноз неблагоприятный. Заболевание может быть диагностировано уже в первом триместре беременности. Исходы и врачебная тактика определяются характером кардиального порока и тяжестью внесердечных аномалий.
Описание. Мы представляем случай трехмесячной выживаемости ребенка из двойни с полной формой пентады Кантрелла. У ребенка при осмотре визуализировался большой дефект передней брюшной стенки, прикрытый тонкими оболочками. Супраумбиликально определялись сокращения эктопированного сердца. Имели место челюстно-лицевые деформации по типу «заячьей губы» и «волчьей пасти». Выражена общая мышечная гипотония. Наличие у ребенка сложного порока сердца явилось противопоказанием к выполнению хирургической коррекции омфалоцеле. В связи с этим была предпринята попытка частичного погружения эвентрированных органов в брюшную полость терапевтическими методами – омфалоцеле с рождения фиксировано за пуповину вертикально (рис. 1 см. на вклейке). В 3 месяца ребенок выписан домой при стабильных показателях гемодинамики с применением кислородного концентратора.
Заключение. В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с использованием современных принципов лечения и выхаживания.
Пентада Кантрелла – редкая врожденная сочетанная патология, характеризующаяся пятью пороками развития: центральным, супраумбиликальным торакоабдоминальным дефектом передней брюшной стенки; дефектом дистального сегмента грудины; эктопией сердца с отсутствием диафрагмального сегмента перикарда; диафрагмальной грыжей; врожденными внутрисердечными аномалиями.
Первое описание этого порока было сделано J.R. Cantrell с соавт. в 1958 году [1], в котором сообщалось о пяти случаях сочетания вышеперечисленных аномалий развития. Полная пентада Кантреллa – тяжелый и редкий, обычно фатальный синдром; чаще встречаются неполные формы, представляющие собой комбинацию двух или трех пороков [2].
В литературе имеется небольшое количество публикаций о данной врожденной патологии. Распространенность варьирует от 5,5 до 7,9 на 1 млн живорожденных [3].
По мнению W.M. Toyama, пентаду Кантрелла делят на 3 класса [4]: 1-й класс (определенный диагноз) – имеются все 5 врожденных пороков, характерных для пентады Кантрелла; 2-й класс (вероятный диагноз) – представлены 4 врожденных дефекта, в том числе пороки сердца и аномалии развития передней брюшной стенки; 3-й класс – неполный вариант, представленный различными комбинациями врожденных дефектов, включающий аномалию грудины.
Этиология пентады Кантрелла неизвестна. Описаны спорадические случаи возникновения заболевания. Чаще страдают мальчики (2,7:1). В литературе описаны единичные случаи сочетания с сиреномелией (синдром русалки), анэнцефалией, синдромом амниотических перетяжек [5]. Описаны семейные случаи изолированной эктопии сердца и пентады Кантрелла [6].
При патологоанатомическом исследовании встречаются определенные виды дефектов: дефект передней брюшной стенки: в 63% случаев – омфалоцеле, у ряда больных описан гастрошизис; стернальные дефекты: расщепление грудины (26%), отсутствие мечевидного отростка (10%), отсутствие нижних двух третей грудины (9%); вентральный ретростернальный дефект диафрагмы в 91% случаев [7]; внутрисердечные аномалии – являются постоянным и обязательным симптомом пентады Кантрелла: большие вентрикулярные септальные дефекты в 100% случаев, атриальные септальные дефекты в 52%, стеноз легочной артерии в 33%, тетрада Фалло в 20% случаев [8]; отсутствие перикарда – в 75% случаев.
Экстракардиальные аномалии включают в себя челюстно-лицевые дефекты, такие как «волчья пасть» и «заячья губа», пороки развития центральной нервной системы: гидроцефалия, анэнцефалия, мальформации скелета: косолапость, отсутствие большеберцовой или лучевой костей, абдоминальные аномалии: полиспления, агенезия желчного пузыря, незавершенный поворот толстой кишки [8, 9].
Наряду с пороками развития описаны хромосомные аберрации [10]. Особое внимание уделяется проведению пренатальной диагностики. Пренатально пентада Кантрелла может быть выявлена при ультразвуковом исследовании уже с 10-й недели беременности. Ультразвуковая диагностика в 2D-реконструкции в первом триместре и дополнительное использование 3D-режима улучшает визуализацию аномалий плода практически в любом его положении [11]. Если по данным ультразвуковой диагностики выставлен диагноз пентада Кантрелла, целесообразно проведение хромосомного анализа, в связи с указаниями на возможную ассоциацию с трисомией, а также с синдромом Тернера [9]. Кроме ультразвуковой диагностики проводится магнитно-резонансная визуализация, которая в сочетании с эхокардиографией плода позволяет провести оптимальную оц.
Ашерова-Юшкова Д.В. , Краснова Л.А. , Яснева Т.З. , Кочешков С.Н. , Лященко А.Ю. , Чапарова Т.В. , Городова Е.В. , Шмелева А.А. , Баданина Ю.С. , Протасова М.О.
Синдром Пена-Шокейра. Пентада Кантрелла
Синдром Пена-Шокейра. Пентада Кантрелла
Синдром Пена-Шокейра (Реnа-Shokeir) является наследственным заболеванием, которое впервые было описано S.D.J. Реnа и М.Н.К. Shokeir в 1974 году. Этот синдром характеризуется артрогрипозом и дисморфическими признаками, возникающими вследствие акинезии плода.
Синонимы. Секвенция акинезии/гипокинезии плода и секвенция фетальных акинетических деформаций.
Этиология. Аутосомно-рецессивная форма наследования заболевания является наиболее распространенной. Имеется несколько описаний необычных проявлений, которые указывают на возможность гетерогенной этиологии.
Риск рецидива. Прогноз риска рецидива бывает неопределен в связи с мультифакториальностью этиологии. В большинстве случаев он находится в пределах от 0 до 25%. Распространенность. Неизвестна.
Патофизиология. Активное шевеление плода начинается с середины первого триместра беременности и имеет основное значение для нормального развития суставов и прилегающих тканей. Отсутствие или уменьшение внутриутробной двигательной активности приводит к тугоподвижности суставов, формированию птеригумов и нарушениям нейромышечных функций со снижением глотательных движений у плода, что вызывает гипоплазию легких и многоводие.
Диагностика. Выявление сочетания патологического положения конечностей, ограниченности движений плода со сниженной или отсутствующей реакцией на акустическую стимуляцию, задержки внутриутробного развития, многоводия и гипоплазии легких позволяет установить диагноз. Также могут выявляться низкое расположение и аномалии ушных раковин, гипертелоризм, короткая шея, расщелина неба, отек мягких тканей головы, деформации грудной клетки, камподактилия и микрогнати. Реже описываемые аномалии при синдроме ПенаШокейра (Pena-Shokeir) включают диафрагмальную грыжу, гастрошизис и микроцефалию.
Дифференциальный диагноз. Трисомия 18 может сопровождаться наличием признаков аналогичных тем, которые обнаруживаются при синдроме Пена-Шокейра (Pena-Shokeir), особенно в отношении черепно-лицевых и внутригрудных аномалий, а также патологии конечностей. Установить диагноз позволяет проведение кариотипирования. Похожее сочетание признаков, за исключением гипоплазии легких, также наблюдается при нелетальных формах артрогрипоза.
Прогноз. Синдром Пена-Шокейра (Pena-Shokeir) является летальным заболеванием. Значительное число пораженных плодов рождается преждевременно. В 30% срочных родов наблюдается мертворождение. Среди выживших большинство детей погибает в течение первых недель жизни. Основной причиной смертности является легочная недостаточность.
Акушерская тактика. Стандартная акушерская тактика ведения беременности изменяется только при возникновении показаний со стороны матери.
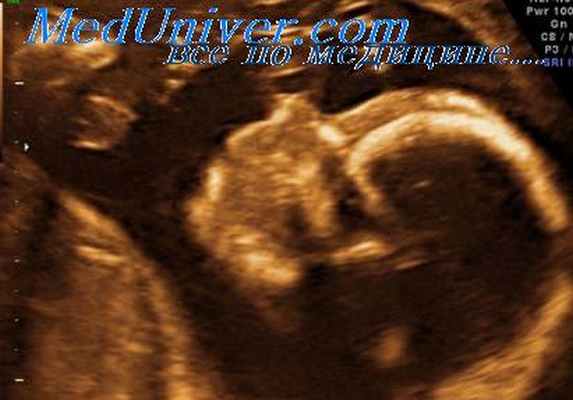
Пентада Кантрелла
Синонимы. Торако-абдоминальная эктопия сердца, эктопия сердца, синдром Кантрелла-Хеллера-Равича (Cantrell-Heller-Ravich); синдром пентады и перитонеоперикардиальная диафрагмальная грыжа.
Сочетанные аномалии. Является правилом наличие внутрисердечных аномалий (таких, как тетрада Фалло (Fallot), дефекты межжелудочковой перегородки). Другие нарушения включают в себя черепно-лицевые аномалии, гидроцефалию, анэнцефалию, незавершенный поворот толстой кишки, косолапость и хромосомные аберрации.
Дифференциальный диагноз. Изолированная торакальная эктопия сердца, эктопия сердца в сочетании с синдромом амниотических перетяжек, аномалия развития стебля тела, изолированное омфалоцеле, синдром Беквита-Видемана (Beckwith-Wiedeman) и хромосомные аномалии.
Прогноз. Выживание является исключительным случаем и зависит от размера дефекта абдоминальной стенки, степени тяжести поражения сердца и наличия сочетанных аномалий. В редких случаях при наличии легких форм возможно проведение хирургической коррекции пороков. В тех ситуациях, когда имеется полная экструзия сердца и органов брюшной полости, прогноз исключительно неблагоприятный.
Акушерская тактика. Родоразрешение рекомендуется проводить в специализированных перинатальных медицинских центрах.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Пентада Кантрелла: клиническое наблюдение
Актульность. Пентада Кантрелла – редкий комбинированный порок развития, представленный пятью признаками: торакоабдоминальным дефектом передней брюшной стенки, дефектами нижней части грудины, передней части диафрагмы и диафрагмального перикарда, а также врожденными аномалиями сердца. Прогноз неблагоприятный. Заболевание может быть диагностировано уже в первом триместре беременности. Исходы и врачебная тактика определяются характером кардиального порока и тяжестью внесердечных аномалий.
Описание. Мы представляем случай трехмесячной выживаемости ребенка из двойни с полной формой пентады Кантрелла. У ребенка при осмотре визуализировался большой дефект передней брюшной стенки, прикрытый тонкими оболочками. Супраумбиликально определялись сокращения эктопированного сердца. Имели место челюстно-лицевые деформации по типу «заячьей губы» и «волчьей пасти». Выражена общая мышечная гипотония. Наличие у ребенка сложного порока сердца явилось противопоказанием к выполнению хирургической коррекции омфалоцеле. В связи с этим была предпринята попытка частичного погружения эвентрированных органов в брюшную полость терапевтическими методами – омфалоцеле с рождения фиксировано за пуповину вертикально (рис. 1 см. на вклейке). В 3 месяца ребенок выписан домой при стабильных показателях гемодинамики с применением кислородного концентратора.
Заключение. В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с использованием современных принципов лечения и выхаживания.
Ключевые слова
Пентада Кантрелла – редкая врожденная сочетанная патология, характеризующаяся пятью пороками развития: центральным, супраумбиликальным торакоабдоминальным дефектом передней брюшной стенки; дефектом дистального сегмента грудины; эктопией сердца с отсутствием диафрагмального сегмента перикарда; диафрагмальной грыжей; врожденными внутрисердечными аномалиями.
Первое описание этого порока было сделано J.R. Cantrell с соавт. в 1958 году [1], в котором сообщалось о пяти случаях сочетания вышеперечисленных аномалий развития. Полная пентада Кантреллa – тяжелый и редкий, обычно фатальный синдром; чаще встречаются неполные формы, представляющие собой комбинацию двух или трех пороков [2].
В литературе имеется небольшое количество публикаций о данной врожденной патологии. Распространенность варьирует от 5,5 до 7,9 на 1 млн живорожденных [3].
По мнению W.M. Toyama, пентаду Кантрелла делят на 3 класса [4]: 1-й класс (определенный диагноз) – имеются все 5 врожденных пороков, характерных для пентады Кантрелла; 2-й класс (вероятный диагноз) – представлены 4 врожденных дефекта, в том числе пороки сердца и аномалии развития передней брюшной стенки; 3-й класс – неполный вариант, представленный различными комбинациями врожденных дефектов, включающий аномалию грудины.
Этиология пентады Кантрелла неизвестна. Описаны спорадические случаи возникновения заболевания. Чаще страдают мальчики (2,7:1). В литературе описаны единичные случаи сочетания с сиреномелией (синдром русалки), анэнцефалией, синдромом амниотических перетяжек [5]. Описаны семейные случаи изолированной эктопии сердца и пентады Кантрелла [6].
При патологоанатомическом исследовании встречаются определенные виды дефектов: дефект передней брюшной стенки: в 63% случаев – омфалоцеле, у ряда больных описан гастрошизис; стернальные дефекты: расщепление грудины (26%), отсутствие мечевидного отростка (10%), отсутствие нижних двух третей грудины (9%); вентральный ретростернальный дефект диафрагмы в 91% случаев [7]; внутрисердечные аномалии – являются постоянным и обязательным симптомом пентады Кантрелла: большие вентрикулярные септальные дефекты в 100% случаев, атриальные септальные дефекты в 52%, стеноз легочной артерии в 33%, тетрада Фалло в 20% случаев [8]; отсутствие перикарда – в 75% случаев.
Экстракардиальные аномалии включают в себя челюстно-лицевые дефекты, такие как «волчья пасть» и «заячья губа», пороки развития центральной нервной системы: гидроцефалия, анэнцефалия, мальформации скелета: косолапость, отсутствие большеберцовой или лучевой костей, абдоминальные аномалии: полиспления, агенезия желчного пузыря, незавершенный поворот толстой кишки [8, 9].
Наряду с пороками развития описаны хромосомные аберрации [10]. Особое внимание уделяется проведению пренатальной диагностики. Пренатально пентада Кантрелла может быть выявлена при ультразвуковом исследовании уже с 10-й недели беременности. Ультразвуковая диагностика в 2D-реконструкции в первом триместре и дополнительное использование 3D-режима улучшает визуализацию аномалий плода практически в любом его положении [11]. Если по данным ультразвуковой диагностики выставлен диагноз пентада Кантрелла, целесообразно проведение хромосомного анализа, в связи с указаниями на возможную ассоциацию с трисомией, а также с синдромом Тернера [9]. Кроме ультразвуковой диагностики проводится магнитно-резонансная визуализация, которая в сочетании с эхокардиографией плода позволяет провести оптимальную оценку комбинации пороков при данной врожденной патологии. Прогноз определяется тяжестью поражения сердца и других сочетанных с ним аномалий.
Описание
Мы представляем пациента с полной формой пентады Кантрелла, находившегося на лечении в отделении реанимации и интенсивной терапии новорожденных и недоношенных детей «Областного перинатального центра» города Ярославля, продолжительность жизни которого составила 3 месяца.
Недоношенный мальчик, массой тела 2650 г, длиной 50 см, окружность головы 34 см, окружность груди 30 см, у женщины 34 лет от 1-й беременности из монохориальной биамниотической двойни, наступившей самопроизвольно. Роды оперативные в сроке 35–36 недель гестации путем экстренного кесарева сечения в связи с плацентарной недостаточностью и страданием 1-го плода. Пренатально у 2-го плода выявлена пентада Кантрелла. В анамнезе хронический пиелонефрит, первичное бесплодие, фетоплацентарная недостаточность 1-го плода. Оценка по шкале Апгар второго ребенка составила 4/5 баллов.
У 1-го ребенка из двойни, мальчика, масса тела которого при рождении составила 1440 г, длина тела 44 см, окружность головы 29,5 см, окружность груди 26 см, оценка по шкале Апгар 6/8 баллов, на фоне задержки внутриутробного развития по гипотрофическому типу постнатально диагностирован врожденный порок сердца – перимембранозный дефект межжелудочковой перегородки 5 мм, два дефекта межпредсердной перегородки 5 и 2 мм, не потребовавшие оперативного лечения. Ребенок выписан из стационара в удовлетворительном состоянии в возрасте одного месяца пяти дней жизни из отделения патологии новорожденных.
Состояние 2-го ребенка из двойни с диагнозом пентада Кантрелла после рождения тяжелое за счет выраженной дыхательной недостаточности, потребовавшей проведения искусственной вентиляции легких в родильном блоке с рождения. Ребенок переведен в отделение реанимации и интенсивной терапии новорожденных для дальнейшего обследования и лечения.
У ребенка при осмотре визуализировался большой дефект передней брюшной стенки, прикрытый тонкими оболочками. Супраумбиликально определялись сокращения эктопированного сердца. Имели место челюстно-лицевые деформации по типу «заячьей губы» и «волчьей пасти». Выражена общая мышечная гипотония.
Результаты лабораторных исследований указывали на отсутствие изменений в гемограмме, умеренное повышение трансаминаз, отрицательные маркеры воспаления. Согласно ультразвуковому исследованию брюшной полости омфалоцеле содержало левую долю печени, поджелудочную железу, кишечные петли, желудок и сердце. Данные компьютерной томографии не дали дополнительной информации.
На рентгенограмме органов грудной клетки отмечалось сужение средостения, образованное сосудистым пучком. Легочные поля не изменены. При риноскопии выявлено значительное искривление носовой перегородки, с фактически полной обтурацией правого носового хода.
Нейросонография указывала на умеренное диффузное повышение эхогенности вещества головного мозга, небольшое расширение ликворо содержащих пространств, высокий индекс резистентности передней и средней мозговых артерий. Данные изменения при ультразвуковом мониторинге с положительной динамикой по показателям мозгового кровотока.
Гастроптоз подтвержден рентгеноконтрастным исследованием. Со стороны органов зрения пороков развития не обнаружено. Проведенное кариотипирование хромосомных аномалий не выявило.
С целью уточнения тактики ведения ребенок был консультирован специалистами ФГБУ Научный центр акушерства, гинекологии и перинатологии им. академика В.И. Кулакова Минздрава России.
Ведущим симптомом в клинической картине являлась дыхательная недостаточность, требовавшая респираторной поддержки в жестких режимах традиционной искусственной вентиляции легких (ИВЛ), с переводом в дальнейшем на осцилляторную высокочастотную вентиляцию в течение двух дней. После чего отмечалась положительная динамика в виде перевода на традиционную ИВЛ с постепенным снижением параметров вентиляции, сменой на неинвазивные методы дыхательной поддержки (CPAP), масочную подачу кислорода в возрасте одного месяца 15 дней жизни. За время нахождения в отделении ребенок перенес аспирационную пневмонию.
Ребенку со сложным пороком сердца потребовалось назначение вазопростана с первых часов жизни для поддержания гемодинамики. Гемодинамические показатели стабилизировались с применением вазопрессорной терапии к 7-м суткам жизни.
Проводилось комбинированное питание: энтеральное – через назогастральный зонд; парентеральное питание через центральный венозный катетер с расчетом всех компонентов – белков, жиров и углеводов по физиологической потребности для данного гестационного возраста. Особое внимание уделялось профилактике дискинезии желудочно-кишечного тракта в связи с наличием пороков развития грудины, диафрагмы и передней брюшной стенки.
Наличие у ребенка сложного порока сердца стало противопоказанием к выполнению хирургической коррекции омфалоцеле. В связи с этим была предпринята попытка частичного погружения эвентрированных органов в брюшную полость терапевтическими методами – омфалоцеле с рождения фиксировано за пуповину вертикально (рис. 1 см. на вклейке). В динамике отмечалось сокращение диаметра дефекта с образованием плотной корки и активная эпителизация по периметру (рис. 2 см. на вклейке).
В возрасте 2 месяцев 12 дней мальчик переведен для дальнейшего лечения в кардиологическое отделение.
Прибавка массы составила 1500 г за три месяца жизни, увеличение длины тела на 5 см за этот период жизни. В неврологическом статусе – с возраста одного месяца жизни фиксировал взгляд, в 2 месяца следил за погремушкой с поворотом головы. В 3 месяца ребенок выписан домой при стабильных показателях гемодинамики с применением кислородного концентратора.
На четвертые сутки пребывания в домашних условиях отмечалось резкое ухудшение состояния за счет нарастания симптомов дыхательной недостаточности. Ребенку вызвана скорая помощь, к моменту приезда которой диагностирована остановка сердца. Проводимые реанимационные мероприятия успеха не имели. Констатирована смерть в возрасте трех месяцев 4 суток жизни.
Пентада Кантрелла представляет собой крайне редкий комбинированный порок развития с неблагоприятным прогнозом. При полной форме данной сочетанной врожденной патологии, особенно при сочетании с аномальным кариотипом рекомендуется прерывание беременности. В случае пренатально диагностированной пентады Кантрелла и при настойчивом желании родителей сохранить ребенка возможно вынашивание беременности с дальнейшей междисциплинарной оценкой консилиумом способа родоразрешения, тактики ведения ребенка в раннем неонатальном периоде, объемом и последовательностью постнатальных вмешательств.
В нашем случае решение о пролонгировании беременности у женщины с длительным бесплодием было продиктовано наличием второго полноценного плода.
Лечебная тактика определяется размерами дефекта передней брюшной стенки, типом эктопии сердца и сочетанными аномалиями [9]. После рождения хирургическое лечение омфалоцеле должно быть незамедлительным. Одновременно должна осуществляться попытка устранения диафрагмального и перикардиального дефектов [12, 13].
Сложности хирургической коррекции у детей с данной патологией зачастую связаны с дефицитом торакоабдоминальных тканей и невозможностью погружения эктопированного сердца в грудную клетку из-за анатомических особенностей. У некоторых пациентов имеется выраженная дыхательная недостаточность, обусловленная гипоплазией легких, осложняющая проведение анестезиологического пособия, течение послеоперационного периода.
Летальность больных со сложной врожденной мальформацией, прооперированных в первые дни жизни превышает 50% [14]. B.S. Norma с соавт. сообщили о благоприятных исходах хирургической коррекции пороков сердца у 22 пациентов с пентадой Кантрелла без его эктопии [13]. L.K. Hornberger с соавт. опубликовали результаты наблюдения и лечения 13 больных пентадой Кантрелла с эктопированным сердцем (4 торакальных и 9 торакоабдоминальных эктопий). Трое из пациентов, с выраженной экстракардиальной патологией, погибли до хирургического вмешательства. Десять больных прооперировано в неонатальном периоде. Продолжительность жизни в пяти случаях составила от 3,5 до 9,5 года. Авторы пришли к выводу, что новорожденные дети с торакальной и торакоабдоминальной эктопией в сочетании с врожденным пороком сердца, при отсутствии клинически значимых экстракардиальных аномалий, имели высокий шанс успешно перенести операцию [15].
В 2008 J.H. Van Hoorn с соавт. представили 58 новорожденных, из которых 33 имели полную и 23 – неполную формы пентады Кантрелла. Двух пациентов классифицировать окончательно не удалось. Четырнадцать больных имели эктопированное сердце без грубых структурных аномалий, 16 – типичное расположение его с внутрисердечным дефектом и 23 – как эктопию, так и врожденный порок сердца. У 29 пациентов присутствовали и другие аномалии, характерные для пентады Кантрелла. 27 из 58 новорожденных умерли в первые сутки после рождения. Смертность была выше в группе детей с полной формой пентады Кантрелла и ассоциированными аномалиями [16].
Заключение
Знание такого редкого заболевания, как пентада Кантрелла специалистами ультразвуковой диагностики, акушерами-гинекологами, неонатологами и детскими хирургами позволяет проводить квалифицированное консультирование женщины с ранних сроков беременности о целесообразности пролонгирования беременности в декретированные сроки, решать вопросы пренатального мониторинга лабораторных и ультразвуковых показателей в условиях многопрофильной акушерской клиники.
Исход и врачебная тактика ведения ребенка с пентадой Кантрелла во многом определяются не только его формой, но и более ранней пренатальной диагностикой аномалии, которая позволяет применять высокотехнологичную медицинскую помощь на всех этапах лечения.
Случай трехмесячной продолжительности жизни ребенка с полной формой пентады Кантрелла, описанный нами, с абдоминальной эктопией сердца, сопутствующими внесердечными аномалиями при отсутствии хирургической коррекции врожденного порока – явление уникальное. Причина смерти нашего больного, вероятнее всего, носит кардиальный характер. Травма, тампонада сердца, внезапная смерть, эндокардит, периферическая эмболия, сердечная недостаточность, аритмия, как осложнения и причина смерти детей с пентадой Кантрелла описаны в литературе [14].
По нашему мнению, значимым фактором, приводящим к летальному исходу в домашних условиях, является прогрессирующая дискинезия желудочно-кишечного тракта (срыгивания, рвота) на фоне диспноэ у детей с врожденными пороками сердца, которая приводит к развитию аспирационной пневмонии, декомпенсации дыхательной и сердечно-сосудистой систем.
В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с использованием современных принципов лечения и выхаживания.
Список литературы
Об авторах / Для корреспонденции
Синдром пена-шокейра. Пентада кантрелла
Синдром Пена-Шокейра (Реnа-Shokeir) является наследственным заболеванием, которое впервые было описано S.D.J. Реnа и М.Н.К. Shokeir в 1974 году. Этот синдром характеризуется артрогрипозом и дисморфическими признаками, возникающими вследствие акинезии плода.
Синонимы. Секвенция акинезии/гипокинезии плода и секвенция фетальных акинетических деформаций.
Этиология. Аутосомно-рецессивная форма наследования заболевания является наиболее распространенной. Имеется несколько описаний необычных проявлений, которые указывают на возможность гетерогенной этиологии.
Риск рецидива. Прогноз риска рецидива бывает неопределен в связи с мультифакториальностью этиологии. В большинстве случаев он находится в пределах от 0 до 25%. Распространенность. Неизвестна.
Патофизиология. Активное шевеление плода начинается с середины первого триместра беременности и имеет основное значение для нормального развития суставов и прилегающих тканей. Отсутствие или уменьшение внутриутробной двигательной активности приводит к тугоподвижности суставов, формированию птеригумов и нарушениям нейромышечных функций со снижением глотательных движений у плода, что вызывает гипоплазию легких и многоводие.
Диагностика. Выявление сочетания патологического положения конечностей, ограниченности движений плода со сниженной или отсутствующей реакцией на акустическую стимуляцию, задержки внутриутробного развития, многоводия и гипоплазии легких позволяет установить диагноз. Также могут выявляться низкое расположение и аномалии ушных раковин, гипертелоризм, короткая шея, расщелина неба, отек мягких тканей головы, деформации грудной клетки, камподактилия и микрогнати. Реже описываемые аномалии при синдроме ПенаШокейра (Pena-Shokeir) включают диафрагмальную грыжу, гастрошизис и микроцефалию.
Дифференциальный диагноз. Трисомия 18 может сопровождаться наличием признаков аналогичных тем, которые обнаруживаются при синдроме Пена-Шокейра (Pena-Shokeir), особенно в отношении черепно-лицевых и внутригрудных аномалий, а также патологии конечностей. Установить диагноз позволяет проведение кариотипирования. Похожее сочетание признаков, за исключением гипоплазии легких, также наблюдается при нелетальных формах артрогрипоза.
Прогноз. Синдром Пена-Шокейра (Pena-Shokeir) является летальным заболеванием. Значительное число пораженных плодов рождается преждевременно. В 30% срочных родов наблюдается мертворождение. Среди выживших большинство детей погибает в течение первых недель жизни. Основной причиной смертности является легочная недостаточность.
Акушерская тактика. Стандартная акушерская тактика ведения беременности изменяется только при возникновении показаний со стороны матери.
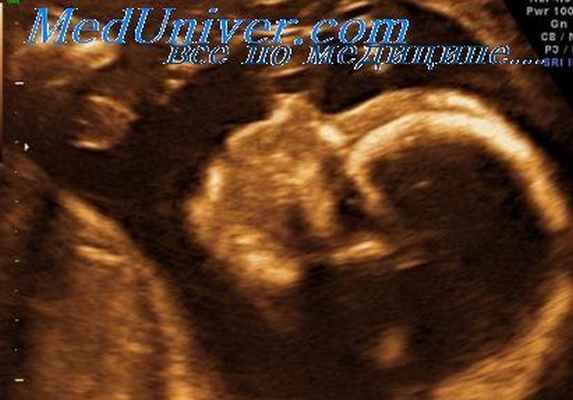
Пентада Кантрелла
Синонимы. Торако-абдоминальная эктопия сердца, эктопия сердца, синдром Кантрелла-Хеллера-Равича (Cantrell-Heller-Ravich)- синдром пентады и перитонеоперикардиальная диафрагмальная грыжа.
Сочетанные аномалии. Является правилом наличие внутрисердечных аномалий (таких, как тетрада Фалло (Fallot), дефекты межжелудочковой перегородки). Другие нарушения включают в себя черепно-лицевые аномалии, гидроцефалию, анэнцефалию, незавершенный поворот толстой кишки, косолапость и хромосомные аберрации.
Дифференциальный диагноз. Изолированная торакальная эктопия сердца, эктопия сердца в сочетании с синдромом амниотических перетяжек, аномалия развития стебля тела, изолированное омфалоцеле, синдром Беквита-Видемана (Beckwith-Wiedeman) и хромосомные аномалии.
Прогноз. Выживание является исключительным случаем и зависит от размера дефекта абдоминальной стенки, степени тяжести поражения сердца и наличия сочетанных аномалий. В редких случаях при наличии легких форм возможно проведение хирургической коррекции пороков. В тех ситуациях, когда имеется полная экструзия сердца и органов брюшной полости, прогноз исключительно неблагоприятный.
Акушерская тактика. Родоразрешение рекомендуется проводить в специализированных перинатальных медицинских центрах.
Синдром піна-шокейра. Пентада кантрелла
Синдром Піна-Шокейра (Реnа-Shokeir) є спадковим захворюванням, яке вперше було описано S.D.J. Реnа і М.Н.К. Shokeir в 1974 році. Цей синдром характеризується Артрогрипоз і дисморфическими ознаками, що виникають унаслідок акинезії плода.
Синоніми. Секвенция акинезії / гіпокінезії плоду і секвенция фетальних акінетіческіх деформацій.
Етіологія. Аутосомно-рецесивна форма успадкування захворювання є найбільш поширеною. Є кілька описів незвичайних проявів, які вказують на можливість гетерогенної етіології.
ризик рецидиву. Прогноз ризику рецидиву буває невизначений у зв`язку з мультифакторіальні етіології. У більшості випадків він знаходиться в межах від 0 до 25%. Поширеність. Невідома.
Патофізіологія. Активне ворушіння плода починається з середини першого триместру вагітності і має основне значення для нормального розвитку суглобів і прилеглих тканин. Відсутність або зменшення внутрішньоутробної рухової активності призводить до тугоподвижности суглобів, формуванню птерігумов і порушень нейром`язових функцій зі зниженням ковтальних рухів у плода, що викликає гіпоплазію легень і багатоводдя.
діагностика. Виявлення поєднання патологічного стану кінцівок, обмеженості рухів плода зі зниженою або відсутньою реакцією на акустичну стимуляцію, затримки внутрішньоутробного розвитку, багатоводдя і гіпоплазії легенів дозволяє встановити діагноз. Також можуть виявлятися низьке розташування і аномалії вушних раковин, гипертелоризм, коротка шия, ущелина неба, набряк м`яких тканин голови, деформації грудної клітки, камподактілія і мікрогнатії. Рідше описувані аномалії при синдромі ПенаШокейра (Pena-Shokeir) включають діафрагмальну грижу, гастрошизис і мікроцефалія.
Диференціальний діагноз. Трисомія 18 може супроводжуватися наявністю ознак аналогічних тим, які виявляються при синдромі Пена-Шокейра (Pena-Shokeir), особливо щодо черепно-лицевих і внутрішньогрудних аномалій, а також патології кінцівок. Встановити діагноз дозволяє проведення каріотипування. Схоже поєднання ознак, за винятком гіпоплазії легенів, також спостерігається при нелетальних формах артрогрипоз.
прогноз. Синдром Піна-Шокейра (Pena-Shokeir) є летальним захворюванням. Значне число уражених плодів народжується передчасно. У 30% термінових пологів спостерігається мертвонародження. Серед тих, що вижили більшість дітей гине протягом перших тижнів життя. Основною причиною смертності є легенева недостатність.
акушерська тактика. Стандартна акушерська тактика ведення вагітності змінюється тільки при виникненні показань з боку матері.
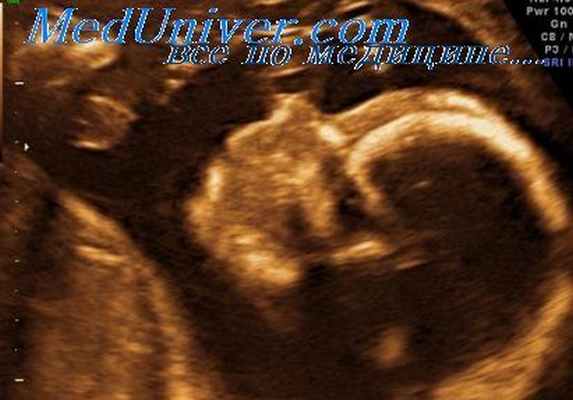
пентада Кантрелла
дане вроджене захворювання характеризується двома основними дефектами: ектопією серця і дефектом передньої черевної стінки (найчастіше спостерігається омфалоцеле, але може зустрічатися і гастрошизис) в поєднанні з порушенням розвитку трьох, пов`язаних між собою структур: дистального відділу грудини, передній частині діафрагми і діафрагмального відділу перикарда. Є повідомлення про варіанти класичної форми, які характеризуються неповним проявом даного синдрому.
Синоніми. Торако-абдомінальна ектопія серця, ектопія серця, синдром Кантрелла-Хеллера-Равича (Cantrell-Heller-Ravich) - синдром пентади і перітонеоперікардіальная диафрагмальная грижа.
поширеність. Зустрічається дуже рідко. У літературі є повідомлення приблизно про 90 клінічних спостереженнях і ще меншому їх числі з повним підтвердженням наявного синдрому.
Етіологія. Невідома. Іноді може поєднуватися з хромосомними аномаліями.
ризик рецидиву. Невідомий. Даних про рецидиви немає. Опубліковано повідомлення про один випадок конкордантность розвитку синдрому у монозиготних близнюків.
діагностика. Наявність повної форми дізрупціі пороку розвитку передніх стінок грудної клітки і черевної порожнини, яка супроводжується п`ятьма ознаками, описаними J.R. Cantrell et al., Є класичною формою пентади і може бути виявлена при ультразвуковому дослідженні починаючи з середини другого триместру. Виявлення скорочується поза межами грудної клітини серця в поєднанні з омфалоцеле дозволяє встановити діагноз пентади. Для виключення хромосомних аномалій рекомендується проведення каріотипування. Внаслідок компресії органів як грудної клітки, так і черевної порожнини можуть виникати асцит і гідроторакс.
поєднані аномалії. Є правилом наявність внутрішньосерцевих аномалій (таких, як тетрада Фалло (Fallot), дефекти міжшлуночкової перегородки). Інші порушення включають в себе черепно-лицьові аномалії, гідроцефалію, аненцефалію, незавершений поворот товстої кишки, клишоногість і хромосомніаберації.
Диференціальний діагноз. Ізольована торакальна ектопія серця, ектопія серця в поєднанні з синдромом амниотических перетяжок, аномалія розвитку стебла тіла, ізольоване омфалоцеле, синдром Беквита-Видемана (Beckwith-Wiedeman) і хромосомні аномалії.
прогноз. Виживання є винятковим випадком і залежить від розміру дефекту абдомінальної стінки, ступеня тяжкості ураження серця і наявності поєднаних аномалій. У рідкісних випадках при наявності легких форм можливе проведення хірургічної корекції вад. У тих ситуаціях, коли є повна екструзія серця і органів черевної порожнини, прогноз виключно несприятливий.
Акушерська тактика. Розродження рекомендується проводити в спеціалізованих перинатальних медичних центрах.
Читайте также: