Синдром Поланда. Секвенция Поттер и синдром prune-belly
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Врожденные пороки развития — далеко не редкость. Они могут быть как слабо, так и сильно выраженными, но в любом случае доставляют значительный физический и психологический дискомфорт. Синдром Поланда — это достаточно редко встречающийся врожденный порок развития, более характерный для мужчин, который в некоторых случаях может вызывать инвалидность (если присутствует множественная деформация тканей, органов, костного скелета). У женщин он встречается не так часто, но, тем не менее, современная медицина разработала несколько вполне эффективных способов для устранения, коррекции видимых дефектов на теле.
Для женщин любого возраста красивое симметричное тело — это обязательное условие психологического комфорта. Синдром Поланда проявляется в явной деформации грудной клетки, ребер, кистей рук, большой и малой грудной мышцы. Особенностью этого врожденного дефекта развития является то, что он не имеет статичной четко определенной выраженности. Соответственно, лечение и способы коррекции подбираются индивидуально в каждом случае после тщательной диагностики пациентов.
Синдром Поланда — причины и проявления
Несмотря на интенсивные темпы развития медицины во всех направлениях, далеко не все заболевания изучены досконально даже при условии наличия нескольких эффективных способов борьбы с ними. Это касается и синдрома Поланда, лечение которого в Украине уже в течение многих лет успешно проводят специалисты Verum Expert clinic. При ярко выраженной клинической картине, причины развития врожденной патологии сегодня до конца не ясны. Общепринятая гипотетическая теория объясняет развитие этого процесса нарушением эмбрионального кровоснабжения и недостаточной интенсивностью кровотока по подключичной и позвоночной артерии на 6 неделе внутриутробного развития.
С недавнего времени также существует другая теория — нарушение миграции эмбриональных тканей в первом триместре. К сожалению, четкого понимания первопричин этого процесса на сегодня нет, что не позволяет предотвратить его развитие, поэтому остается лишь бороться с последствиями. Они выражаются следующим образом: аплазия;
- гипоплазия;
- килевидная деформация грудной клетки;
- воронкообразная деформация грудной клетки;
- отсутствие подкожного жирового слоя на грудине;
- брахиосиндактилия;
- алопеция.
Сегодня синдром Поланда — это не приговор и не повод мириться с врожденными дефектами, ведь его эффективное комплексное лечение в городе Киев (Украина) проводят специалисты Verum Expert clinic.
Синдром Поланда у женщин — сколько стоит операция в Киеве?
Как и в случае с другими врожденными дефектами развития, цена лечения определяется индивидуально. Она вариабельна во многом потому, что выраженность синдромов у всех пациентов разная, как и их набор. Именно эти факторы определяют, какой вид лечения необходимо применять и каково будет предполагаемое количество, сложность комплекса восстановительных процедур. Пациентам могут потребоваться следующие виды хирургического вмешательства:
- восстановление мышечной ткани, жирового подкожного слоя на груди;
- имплантация молочных желез;
- формирование соска;
- устранение деформации грудины;
- восстановление костно-хрящевой структуры ребер;
- устранение дефектов кистей, пальцев рук.
- создание мускульного каркаса для имплантации.
Verum Expert clinic — это современная клиника с новейшей материально-технической базой и лучшими компетентными специалистами, которые могут оказать помощь даже в самых сложных случаях. Все пациенты обязательно проходят первичную консультацию, осмотр у врача и полную диагностику для определения клинической картины и подбора оптимального лечения. Узнать стоимость услуг и записаться на прием к специалисту можно на сайте verum.ua.
Синдром Поланда. Секвенция Поттер и синдром prune-belly
Синдром Поланда. Секвенция Поттер и синдром prune-belly
Односторонняя симбрахидактилия и ипсилатеральная аплазия стернальной порции большой грудной мышцы. У женщин также отмечается ипсилатеральная аплазия молочной железы. В нескольких случаях сообщалось о декстракардии.
Синонимы. Синдром Поланда-Мебиуса (Poland-Moebius) и секвенция прекращения кровоснабжения из подключичной артерии.
Распространенность. Частота встречаемости составляет 0,2 на 10 000 родов.
Этиология. Спорадическое прекращение кровоснабжения из подключичных артерий у эмбриона наранних стадиях развития. Описано несколько семейных случаев.
Диагностика. Может быть выявлена асимметрия грудной клетки и симбрахидактилия, однако патология длинных трубчатых костей, вероятно, настолько незначительна, что практически не определяется.
Дифференциальный диагноз. При выявлении одностороннего характера аномалий существует лишь несколько дифференциальных диагнозов.
Прогноз. За исключением особенностей, связанных с наличием патологии конечностей прогноз очень благоприятный.
Акушерская тактика. Изменения стандартной пренатальной тактики ведения не требуется. Важное значение для генетического консультирования семейной пары имеет подтверждение диагноза после рождения.
Секвенция Поттер
Эпоним Поттер (Potter) используется в названии нескольких заболеваний.
1. Синдром Поттер (Potter). В настоящее время называется также секвенция маловодия или двухсторонняя агенезия почек в зависимости от того, относится ли это к причине синдрома (вторая ситуация) или к его механизму (первая ситуация).
2. Синдром Поттер (Potter), тип I. В настоящее время называется аутосомно-рецессивной поликистозной болезнью почек.
3. Синдром Поттер (Potter), тип II. В настоящее время называется мультикистозной дисплазией почек.
4. Синдром Поттер (Potter), тип III. В настоящее время называется аутосомно-доминантной поликис-тозной болезнью почек.
Синдром prune-belly
Синдром prune-belly (также может быть переведен как синдром недостаточности мышц живота, синдром «подрезанного» живота или живота «в виде чернослива». - Примеч. ред.) является редко встречающимся врожденным заболеванием, более распространенным у лиц мужского пола, и проявляется дефектом (аплазией или гипоплазией) развития мышц передней брюшной стенки, крипторхизмом и аномалиями мочеполовой системы.
Синонимы. Синдром Игла-Баррета (Eagle-Barret).
Этиология. Для объяснения патогенеза данного заболевания было предложено много теорий:
• аномалии развития мезенхимы: остановка развития элементов мезенхимы приводит к выраженному растяжению мышц живота и дефектам развития мочевыводящего тракта;
• первичная обструктивная аномалия мочевыво-дящих путей: обструкция в области устья мочевого пузыря в сочетании с обструкцией мочеиспускательного канала, что приводит к перерастяжению передней брюшной стенки, вызывая развитие синдрома prun-belly;
• генетический дефект: подозревается вследствие преобладания у лиц мужского пола и наблюдений нескольких семейных случаев
• перерастяжение брюшной стенки, вызываемое множественными факторами.
Распространенность. От 0,25 до 0,3 на 10 000 новорожденных, почти исключительно у мальчиков. Риск рецидива. Неизвестен, но низок.
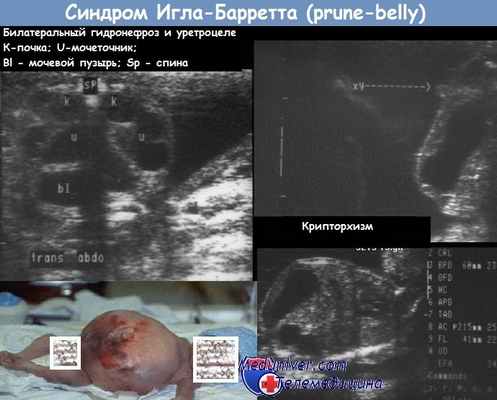
Диагностика. Диагноз должен предполагаться у плодов, имеющих большие образования в брюшной полости. Их наличие обычно бывает обусловлено обструкцией выхода из мочевого пузыря (уретральные клапаны или агенезия уретры). Кроме того, причиной могут являться другие большие объемные образования в брюшной полости, такие как киста яичника и гидрометрокольпос. Обнаружение крипторхизма обычно бывает невозможно из-за выраженного маловодия.
Дифференциальный диагноз. Дифференциальный диагноз проводится с аномалиями мочевыводящего тракта, такими как мегацистис и мегауретер, обструкция уретры и первичный пузырно-мочеточниковый рефлюкс. Также могут рассматриваться нейрогенный мочевой пузырь и синдром мегацистиса - микроколона - гипоперистальтики кишечника, хотя в этом случае будет обнаруживаться нормальное количество околоплодных вод.
Прогноз. Зависит от степени выраженности нарушения функции почек. Ранняя обструкция, встречающаяся чаще всего, приводит к почечной недостаточности, гипоплазии легких и гибели в неонатальном периоде. Ранняя декомпрессия при выраженной обструкции оттока из мочевого пузыря с помощью везикоамниотического шунтирования может предотвратить или смягчить эти осложнения, тем самым улучшая прогноз. Плоды, у которых развивается незначительное расширение мочевыводящих путей, имеют лучший прогноз. В таких случаях единственными проявлениями могут быть незначительно выраженный гидронефроз и мегауретер.
Акушерская тактика. В течение всей беременности требуется проводить эхографический мониторинг состояния мочевыводящих путей и оценку объема околоплодных вод. В определенных случаях предлагалось проведение ранней декомпрессии мочевого пузыря для улучшения функции почек и легких, если обнаруживалась ранняя или тяжелая степень расширения мочевыводящих путей. В тяжелых случаях после рождения может потребоваться трансплантация почек.
- Вернуться в оглавление раздела "Акушерство."
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром поланд. Секвенция поттер і синдром prune-belly
одностороння сімбрахідактілія і Ипсилатеральная аплазія стернальной порції великого грудного м`яза. У жінок також відзначається Ипсилатеральная аплазія молочної залози. У кількох випадках повідомлялося про декстракардія.
Синоніми. Синдром Поланд-Мебіуса (Poland-Moebius) і секвенция припинення кровопостачання з підключичної артерії.
поширеність. Частота народження становить 0,2 на 10 000 пологів.
Етіологія. Спорадичне припинення кровопостачання з підключичних артерій у ембріона наранніх стадіях розвитку. Описано кілька сімейних випадків.
діагностика. Може бути виявлена асиметрія грудної клітки і сімбрахідактілія, проте патологія довгих трубчастих кісток, ймовірно, настільки незначна, що практично не визначається.
Диференціальний діагноз. При виявленні одностороннього характеру аномалій існує лише кілька диференціальних діагнозів.
прогноз. За винятком особливостей, пов`язаних з наявністю патології кінцівок прогноз дуже сприятливий.
акушерська тактика. Зміни стандартної пренатальної тактики ведення не потрібно. Важливе значення для генетичного консультування сімейної пари має підтвердження діагнозу після народження.
секвенция Поттер
епонім Поттер (Potter) використовується в назві декількох захворювань.
1. Синдром Поттер (Potter). В даний час називається також секвенция маловоддя або двостороння агенезія нирок в залежності від того, чи стосується це спричинило причини синдрому (друга ситуація) або до його механізму (перша ситуація).
2. Синдром Поттер (Potter), тип I. В даний час називається аутосомно-рецесивною полікістозній хворобою нирок.
3. Синдром Поттер (Potter), тип II. В даний час називається Мультікістозная дисплазією нирок.
4. Синдром Поттер (Potter), тип III. В даний час називається аутосомно-домінантною полікіс-тозние хворобою нирок.
Синдром prune-belly
Синдром prune-belly (Також може бути переведений як синдром недостатності м`язів живота, синдром «подрезанного» живота або живота «у вигляді чорносливу». - Прим. Ред.) Є рідкісним вродженим захворюванням, більш поширеним в осіб чоловічої статі, і проявляється дефектом (аплазією або гіпоплазією ) розвитку м`язів передньої черевної стінки, крипторхізм і аномаліями сечостатевої системи.
Синоніми. Синдром Голка-Баррета (Eagle-Barret).
Етіологія. Для пояснення патогенезу даного захворювання було запропоновано багато теорій:
• аномалії розвитку мезенхіми: зупинка розвитку елементів мезенхіми призводить до вираженого розтягування м`язів живота і дефектів розвитку сечовивідного тракта-
• первинна обструктивна аномалія сечовивідних-дящих шляхів: обструкція в області гирла сечового міхура в поєднанні з обструкцією сечівника, що призводить до перерозтягнення передньої черевної стінки, викликаючи розвиток синдрому prun-belly-
• генетичний дефект: підозрюється внаслідок переважання у осіб чоловічої статі і спостережень декількох сімейних випадків
• перерозтягнення черевної стінки, що викликається множинними факторами.
поширеність. Від 0,25 до 0,3 на 10 000 новонароджених, майже виключно у хлопчиків. Ризик рецидиву. Невідомий, але низький.
діагностика. Діагноз повинен передбачатися у плодів, що мають великі освіти в черевній порожнині. Їх наявність зазвичай буває обумовлено обструкцією виходу з сечового міхура (уретральні клапани або агенезія уретри). Крім того, причиною можуть бути інші великі об`ємні освіти в черевній порожнині, такі як кіста яєчника і гідрометрокольпос. Виявлення крипторхізму зазвичай буває неможливо через вираженого маловоддя.
Диференціальний діагноз. Диференціальний діагноз проводиться з аномаліями сечовивідного тракту, такими як мегацістіс і мегауретер, обструкція уретри і первинний міхурово-сечовідний рефлюкс. Також можуть розглядатися нейрогенний сечовий міхур і синдром мегацістіса - мікроколонії - гіпоперістальтікі кишечника, хоча в цьому випадку буде виявлятися нормальна кількість навколоплідних вод.
прогноз. Залежить від ступеня вираженості порушення функції нирок. Рання обструкція, що зустрічається найчастіше, приводить до ниркової недостатності, гіпоплазії легенів і загибелі в неонатальному періоді. Рання декомпресія при вираженій обструкції відтоку з сечового міхура за допомогою везікоамніотіческого шунтування може запобігти або пом`якшити ці ускладнення, тим самим покращуючи прогноз. Плоди, у яких розвивається незначне розширення сечовивідних шляхів, мають кращий прогноз. У таких випадках єдиними проявами можуть бути незначно виражений гідронефроз і мегауретер.
акушерська тактика. Протягом всієї вагітності потрібно проводити ехографіческій моніторинг стану сечовивідних шляхів і оцінку обсягу навколоплідних вод. У певних випадках пропонувалося проведення ранньої декомпресії сечового міхура для поліпшення функції нирок і легень, якщо виявлялася рання або важка ступінь розширення сечовивідних шляхів. У важких випадках після народження може знадобитися трансплантація нирок.
Синдром меккеля. Діагностика і прогноз при синдромі меккеля
це рідко зустрічається летальний синдром, який характеризується потиличних цефалоцеле, постаксіальной полідактилія і кістозної дисплазією нирок. Він може поєднуватися з багатьма іншими станами, найбільш поширеним з яких є фіброз печінки.
Синоніми. Дісенцефалія з кістозними змінами внутрішніх органів і синдром Меккеля (Meckel) (прийнятий в англомовній літературі). Синдром Меккеля (Meckel) є кращим назвою захворювання і використовується як при пошуку в інтернет-базі даних Medline, так і в Британській енциклопедії вроджених вад (Birth Defect Encyclopedia). Іншими назвами є синдром Грубера (Gruber) (використовується в європейській літературі) і синдром Меккеля-Грубера (Meckel-Gruber).
поширеність. Точно невідома, проте по загальноприйнятій думці це дуже рідко зустрічається патологія. За даними D. Bergsma, поширеність синдрому Меккеля (Meckel) становить 0,2 на 10 000 новонароджених. R. Salonen і R. Norio встановили, що частота даного синдрому при народженні варіює від 0,07 до 0,7 на 10 000 пологів.
генетичні порушення. Локус, відповідальний за виникнення синдрому Меккеля (Meckel), локалізована на довгому плечі хромосоми 17q2.1-q2.4. Фенотипическая варіабельність і випадки без підтвердженої зв`язку з хромосомою 17q припускають наявність деякої міри локусной гетерогенності.
діагностика. У 1981 році F.C. Fraser і A. Lytwyn припустили, що кістозна дисплазія нирок є постійною аномалією при синдромі Меккеля (Meckel), і тому для встановлення діагнозу вона повинна виявлятися в поєднанні з не менш ніж двома меншими дефектами. Дана концепція продовжує обговорюватися, при цьому поширеність аномалій нирок при даному синдромі становить від 95 до 100%. Нирки спочатку мають мікроскопічні кісти, які розвиваються, руйнуючи її паренхіму і викликаючи збільшення органу в 10-20 разів.
Першим ехографіческім ознакою в більшості випадків є маловоддя внаслідок дисфункції нирок, що виникає на початку другого триместру вагітності, коли продукція сечі повинна заміщати позаклітинне дифузію в якості основного джерела навколоплідних вод. Однак в деяких випадках синдрому може виявлятися нормальна кількість амніотичної рідини, і, таким чином, її наявність не виключає діагнозу. У ряді ситуацій сечовий міхур не буде візуалізуватися. Відсутність ехографічних ознак захворювання на ранніх термінах у вагітної з групи ризику по рецидиву захворювання у плода не виключає синдрому Меккеля (Meckel), і в цих випадках рекомендується спостереження в динаміці до 20 тижнів гестації.
потиличний цефалоцеле зустрічається в 60-80%. У зв`язку з тим, що цефалоцеле оточене мембраною, показники рівня АФП в крові матері або в амніотичної рідини можуть бути нормальними. Постаксіальной полідактилія зустрічається в 55-75%. Також можуть бути присутніми інші аномалії кінцівок, у вигляді їх викривлення і вкорочення. Виявлення, принаймні, двох з трьох ознак класичної тріади при наявності нормального каріотипу дозволяє встановити діагноз.
Диференціальний діагноз. Диференціальний діагноз буде залежати від типу поєднаних аномалій. Внаслідок подібності кількох ехографічних ознак трисомії 13 слід виключити шляхом каріотипування. Іншим можливим захворюванням є аутосомно-домінантна поликистозная хвороба нирок.
поєднані аномалії. Перелік можливих аномалій, що поєднуються з даними синдромом, є великим. У деяких ситуаціях така широка фенотипова варіабельність значно ускладнює встановлення діагнозу.
прогноз. Синдром Меккеля (Meckel) є летальним захворюванням. У більшості випадків відзначаються мертвонародження або рання неонатальна загибель протягом декількох годин або днів після народження. У деяких спостереженнях описано виживання протягом декількох місяців, але з низькою якістю життя. Н.М. Ramadani і Н.А. Nasrat повідомили про дитину, яка померла у віці 28 місяців. У 1997 році P. Paavola et al. привели спостереження ще одного нетипового випадку виживання протягом 18 місяців.
акушерська тактика. При підозрі на синдром Меккеля (Meckel) необхідно провести каріотипування з метою виключення хромосомних захворювань. Якщо приймається рішення про її пролонгацію або якщо діагноз встановлений в більш пізні терміни, стандартна акушерська тактика ведення вагітної не змінюється.
Введение
Синдром Поланда является редким врожденным заболеванием, популяционная частота которого составляет 1 случай на 30–100 тыс. человек, соотношение мужского пола к женскому достигает 2:1 – 3:1. В 75 % случаев синдром Поланда встречается справа.
Синдром Поланда представляет собой комплекс врожденных дефектов, включающий отсутствие кортикостернальной части большой и малой грудных мышц, синдактилию, брахидактилию, ателию и/или амастию, отсутствие реберных хрящей или нескольких ребер (как правило 2-5), отсутствие волос в подмышечной впадине и снижение толщины подкожно-жирового слоя (2,12). Кроме наличия эстетического дефекта в области грудной клетки, синдром Поланда часто сопровождается астмой, болью, нарушением работы сердечно-сосудистой и дыхательных систем, ограничением движений, частыми инфекционными заболеваниями нижних дыхательных путей (20,21). Отдельные симптомы этого синдрома одним из первых описал Lallemand L.M. (1826) и Frorier R. (1839). Однако, назван он по имени английского студента-медика Alfred Poland, который в 1841 году опубликовал более детальное описание данной патологии. Полную характеристику синдрома в литературе впервые опубликовал Thompson J. в 1895 году (14).
Синдром Поланда характеризуется значительным полиморфизмом и все его проявления редко встречаются у одного пациента. J. Thomson в 1895 году и Bing в 1902 году подробно описали недоразвитие большой грудной мышцы и перепончатую одностороннюю синдактилию. Синдактилия является наиболее частым признаком, сопутствующим дефектам развития тканей грудной клетки при данной патологии, в связи с чем в 1962 году Clarkson предложил термин “Синдактилия Поланда” (14). Позже Lauros описал, что у пациентов с синдромом Поланда отсутствует грудинно-реберная часть большой грудной мышцы и отмечается гипоплазия малой грудной мышцы, аномалии развития ребер, и, в редких случаях, грыжа легкого.
Меньше чем у 1% пациентов при синдроме Поланда имеется семейный анамнез по доминантному аутосомному признаку (15,16). Причиной развития может быть прерывание или уменьшение кровотока из грудной артерии или одной из ее периферических ветвей в течение шестой недели беременности. В зависимости от сроков и интенсивности нарушения кровоснабжения синдром Поланда имеет различную степень тяжести (14,15). Так, в литературе подробно описано клиническое наблюдение пациента с синдромом Поланда, у которого присутствовала ателия, при этом была нормально развита большая грудная мышца и лишь недоразвитие передней зубчатой мышцы. Вышеперечисленное было выявлено по данным электромиографии (14).
Методы коррекции эстетического дефекта при синдроме Поланда основываются на желании создать симметрию путем увеличения объема тканей с пораженной стороны. Одними из первых были предложены методы установки силиконового эндопротеза или же экспандера с последующей заменой его на эндопротез (22,23,24).
При отсутствии сосудистой патологии с пораженной стороны, использовали ротационные лоскуты с осевым кровообращением (17). Для восполнения объема недоразвитой или вообще отсутствующих малой и большой грудных мышц, выполняют перемещение лоскута из широчайшей мышцы спины (ШМС) в позицию большой грудной мышцы на сосудисто-мышечной ножке. Если синдрому Поланда сопутствуют сосудистые нарушения с пораженной стороны, это может стать показанием для использования свободной пересадки лоскута широчайшей мышцы спины с контралатеральной стороны(1), пересадки верхнего и нижнего ягодичных лоскутов (9) или в редких случаях использованию DIEP(7). Необходимо учитывать, что при развороте широчайшей мышцы спины, со временем развивается ее атрофия (11). Проблема обширных рубцов в донорской области разрешима при использовании эндоскопической видеотехники (6,8).
Наименее трудоемкий способ коррекции с помощью только силиконового протеза, в последующем имел осложнения в виде дислокация, протрузии и деформации связанной с резорбцией ребер (3).
В 1998 году для коррекции выраженной гипомастии и аплазии грудных мышц, адекватного укрытия импланта была использована методика мобилизации сальника. Хотя за последние годы эта методика была неоднократно модифицирована, трудоемкость и многочисленные осложнения в виде нагноения, атрофии тканей, не позволяют в полной мере применять данный метод(18, 19).
Некоторые авторы, при сохранении грудинно – ключичной части большой грудной мышцы, предлагают ее перемещение для устранения дефекта подмышечного изгиба и одномоментную установку силиконового импланта (13). Использование эндоскопической техники для разворота широчайшей мышцы спины и одномоментное эндопротезирование позволяет избежать обширных рубцов в донорской области (25,26 ).
У мужчин с синдромом Поланда коррекция достигается липосакцией или только силиконовым имплантом со стороны поражения (10).
Современный подход к проблеме коррекции дефекта мягких тканей при синдроме Поланда подразумевает использование аутожира (4) и силиконовых имплантов (5).
Несмотря на существование многочисленных методик, ни одна из них не может удовлетворить всем желаниям хирурга и пациентов, каждая имеет свои недостатки и преимущества. На сегодняшний день использование ТДЛ в сочетание с одномоментным или отсроченным эндопротезированием обеспечивает оптимальный устойчивый эстетических результат. Достижение симметрии является одной из самых главных задач при устранении дефектов, обусловленных синдромом Поланда.
Материалы и методы
Мы выполнили реконструкцию молочной железы при синдроме Поланда у 12 пациенток, у 4 из них (33,4%) наблюдали левосторонний синдром Поланда, у 8 (66,6%) правосторонний. У 8 (67%) пациенток наряду отсутствием молочной железы встречалось недоразвитие верхней конечности, у 3 (25%) пациенток гипоплазия 3,4,5 ребер, у 1 (8%) атрезия ребер. Из 12 пациенток у 8 (66,6%) мы наблюдали полное отсутствие не только малой и большой грудных мышц, но и ткани молочной железы. У 1 (8,3%) пациенток на фоне отсутствия грудных мышц наблюдали незначительный объем молочной железы (размер А), что позволяло использовать для реконструкции только участок широчайшей мышцы спины без кожного островка, и 1 (8,3%) это позволило использовать только эндопротез. У 4 (33,6%) пациенток из 12 была выполнена реконструкция молочной железы с помощью одномоментного разворота торакодорсального лоскута и установки эндопротеза. У 6 (50%) пациенток одномоментно был выполнен разворот широчайшей мышцы спины и установка экспандера с последующей его заменой на эндопротез (Рисунок №1).
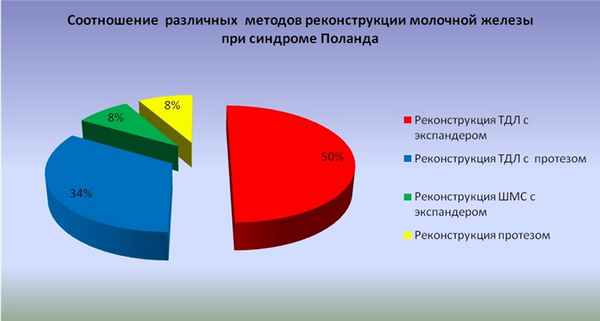
Рис. №1. Частота использования различных способов реконструкции молочной железы при синдроме Поланда
Из 12 пациенток, у 5 (41,6%) мы выполнили для достижения симметрии после коррекции синдрома Поланда редукционную маммопластику, у 7 относительная симметрия достигалась только путем реконструкции дефектной железы.
Во всех клинических наблюдениях отмечали асимметрию САК, но только 2 пациентки обратились с просьбой восстановить симметричность САК.
Хирургическая тактика
Основным фактором, определяющим хирургическую тактику при синдроме Поланда, является степень дефицита тканей в области реконструируемой молочной железы и выбор оптимального способа коррекции.
Отсутствие ткани молочной железы на фоне недоразвития или аплазии грудных мышц в большинстве случаев приводит к необходимости использовать для реконструкции кожно-мышечный лоскут.
Ротация лоскутов на основе широчайшей мышцы спины с кожным с островком (ТДЛ) или без него (ШМС), является наиболее приемлемыми. С помощью этой методики мы избегаем осложнений, связанных со свободной пересадкой лоскута, облегчаем послеоперационный период, к тому же формируется легко скрываемый рубец.
Показания для использования полноценного торакодорсального лоскута (ТДЛ) является не только аплазия или недоразвитие большой и малой грудных мышц, но и полное отсутствие ткани молочной железы (размер 0). Наличие ткани молочной железы (размер А и более) позволяет использовать только лоскут на основе широчайшей мышцы спины (ШМС) без кожного компонента.
В некоторых клинических наблюдениях, когда объем контрлатеральной молочной железы превышал объем С, мы ротировали участок ШМС и устанавливали экспандер и только после получения необходимого объема заменяли его на эндопротез. Безусловно, при выборе хирургической тактики мы учитывали желание пациентки на определенный размер груди и отношение ее к наличию рубцов в донорской и реципиентной зоне.
После достижения желаемого результата с пораженной стороны, при необходимости, мы выполняем редукционную маммопластику или мастопексию с контрлатеральной стороны. Окончательным этапом является формирование сосково-ареолярно комплекса на реконструированной молочной железе – воссоздание соска местными тканями и формирование ареолы с помощью татуажа или свободной пересадки кожи.
Таким образом, можно выделить несколько основных этапов при коррекции синдрома Поланда.
- Устранение дефекта мягких тканей путем с использованием различные видов реконструкции:
– при мышечном дефекте в сочетании с дефицитом кожи, ротирование ТДЛ с одномоментной установкой протеза или экспандера;
– при мышечном дефекте с достаточным кожи и ткани молочной железы, возможно применение ШМС с эедопротезом или только эндопротеза. - Коррекция контрлатеральной молочной железы.
- Реконструкция САК.
Клинический пример 1
Пациентка З., 18 лет, поступила с диагнозом: синдром Поланда справа. Жалобы при поступлении на контурную деформацию грудной клетки справа и недоразвитие молочной железы.
При осмотре на момент поступления: имеется асимметрия молочных желез за счет недоразвития правой молочной железы, кожа над ней обычного цвета, сосково-ареолярный комплекс уменьшен в размере, обычного цвета. Местно: большая грудная мышца справа не пальпируется, пальпация правой молочной железы безболезненная. Левая молочная железа здоровая, соответствует размеру В, при пальпации безболезненная, мягко-эластичной консистенции, без патологических образований.
Основные этапы операции
После предварительной разметки, в проекции широчайшей мышцы спины. Выполняли основной этап забор и разворот участка широчайшей мышцы спины (Рисунок № 2,3).
Рис. №2. Этап забора лоскута
Затем ротировали и размещали мышечный лоскут в область передней стенки грудной клетки (Рисунок №4).
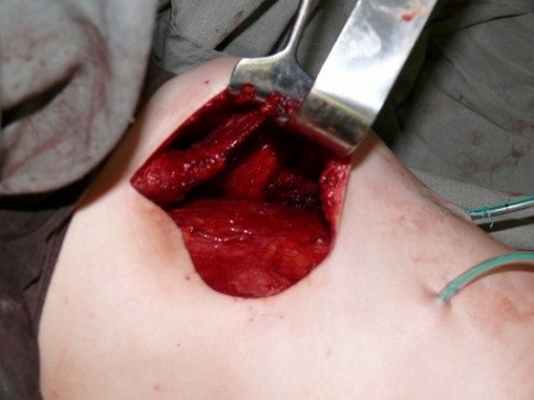
Рис. №4. Карман с ротированной мышцей
В сформированный карман поместили эндопротез (Рисунок №5).
Рис. №5. Субмускулярный карман с эндопротезом Рис. №6. Окончательный вид реконструированной правой молочной железы
В некоторых клинических наблюдениях даже сохранение незначительной асимметрии соответствует требованиям пациентки, хотя хирург предполагает возможность оптимальной симметрии. Данной пациентке (Рисунок №7а) был установлен протез круглой формы размером 325сс (Рисунок 7б,в)
В положении пациентки, стоя «руки за голову» мы отметили приближенную к идеальному симметрию. Хотя на первом этапе нам не удалось достичь полной симметрии, от дальнейшей коррекции асимметрии пациентка отказалась.
Клинический пример 2
Пациентка О., 19 лет. Диагноз: синдром Поланда слева.
Из анамнеза с детства отмечает контурную деформацию грудной клетки слева, с периода полового созревания недоразвитие молочной железы.
Коррекция синдрома Поланда у данной пациентки заняло несколько этапов:
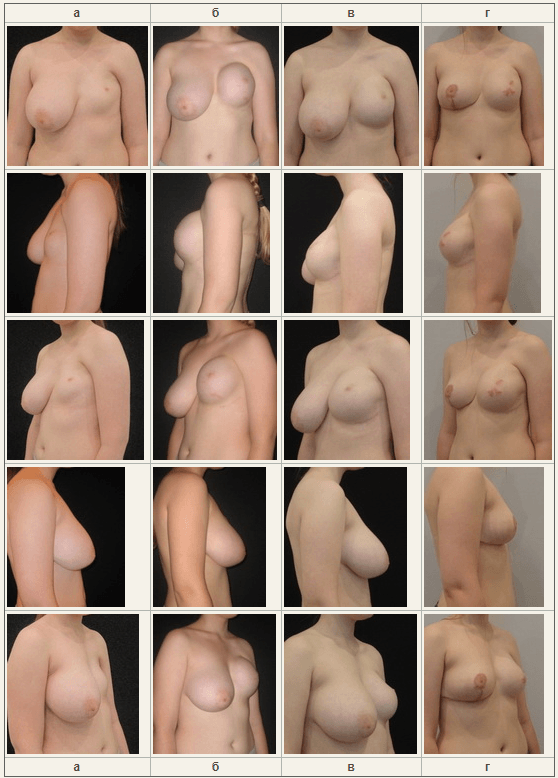
- разворот ШМС мы выполнили разворот ШМС и установку экспандера объемом 400сс;
- замена экспандера на протез анатомической формы объемом 355сс через 6 месяцев;
- редукционная маммопластика правой молочной железы;
- реконструкция САК (Рисунок №8). Рис. №8. “а” – до операции; “б” – после первого этапа, разворота ШМС и установки экспандера; “в” – замена экспандера на эндопротез; “г” – через 3 месяца редукционной маммопластики справа
На наш взгляд, у данной пациентки мы достигли оптимально возможного эстетического результата.
Таким образом, восстановление симметрии при синдроме Поланда – это многоэтапная процедура. Основной задачей является создание адекватного кармана для эндопротеза с достаточным количеством покровных тканей. Выбор хирургической тактики зависит от анатомических данных пациентки – степень поражения мягких тканей, размер и форма здоровой молочной железы.
Читайте также: