Синдром Сильвера-Рассела - клиника, диагностика
Добавил пользователь Евгений Кузнецов Обновлено: 30.01.2026
Синдром Сильвера — Рассела — это наследственное заболевание, проявляющееся внутриутробной задержкой развития в сочетании с низкорослостью и другими стигмами дизэмбриогенеза. В статье представлено описание этой редкой генетической патологии, выявленной у 7-месячного ребенка. Описана динамика физического и неврологического статуса, результаты клинического и лабораторно-инструментального обследования, особенности ведения пациентки Подозрение на генетическую патологию возникло лишь при втором посещении кабинета катамнеза, ближе к 7 мес. жизни, в связи с отставанием в физическом развитии и имеющимися стигмами дизэмбриогенеза. В связи с очень несимметричными пропорциями тела (большая голова относительно тела) в возрасте 5 мес предположили наличие гидроцефалии, которая была отвергнута на основании данных нейросонографии Окончательный диагноз подтвержден результатами генетического исследования, выявившего аномальное метилирование гена Н19 методом микросателлитного анализа локуса 7-й хромосомы.
Ключевые слова: синдром Сильвера — Рассела, ребенок, задержка внутриутробного развития, псевдогидроцефалия, карликовость, генетическое консультирование.
N.R. Khafizova, D.R. Merzlyakova, Yu.F. Safina
Bashkir State Medical University, Ufa, Russian Federation
Russel – Silver syndrome (RSS) is a hereditary disease manifesting with intrauterine growth retardation, dwarfism, and other stigmas of embryopathy. We describe this rare genetic condition in a 7-month-old baby. The changes in physical condition and neurological status, clinical signs, laboratory tests, and management strategy are addressed. A genetic condition was suggested through an arrested development and stigmas of embryopathy only at the age of 7 months. At the age of 5 months, hydrocephaly was suspected due to asymmetrical proportions of the body (the relatively large size of head compared to a small body). However, neurosonography ruled out this diagnosis. Genetic testing for microsatellite loci on chromosome 7, which identified abnormal methylation of H19 gene verified the final diagnosis.
Keywords: Russel – Silver syndrome, child, intrauterine growth retardation, pseudohydrocephalus, dwarfism, genetic counseling.
Введение
В качестве примера приводим описание катамнестического наблюдения ребенка с ССР в условиях ГБУЗ РДКБ г. Уфы.
Клиническое наблюдение
В 5 мес. мама больной девочки обратилась в кабинет катамнеза с жалобами на задержку физического и моторного развития ее ребенка.
Из анамнеза: ребенок от матери 22 лет, семья полная, проживает в одном из районов Республики Башкортостан, материально-бытовые условия удовлетворительные. Бесплодие, не поддающееся лечению. Данная беременность первая, наступила в результате экстракорпорального оплодотворения, протекала на фоне фетоплацентарной недостаточности и плацентарных нарушений 2-й степени. Роды оперативные, на сроке 34 нед.
При рождении выставлен диагноз: задержка внутриутробного развития 3-й степени, симметричная форма
Интранатальная асфиксия с оценкой по шкале Апгар 4/5 баллов. Масса тела при рождении 1110 г, рост 36 см. Сразу после рождения проводились реанимационные мероприятия в связи с дыхательными нарушениями. Девочка была переведена на аппаратное дыхание, выхаживалась в кувезе, получала зондовое кормление грудным материнским молоком. Первые 11 дней жизни ребенок провел в отделении реанимации новорожденных с диагнозом: врожденная пневмония на фоне пневмопатии. Недоношенность 34 нед. Задержка внутриутробного развития 3-й степени по гипопластическому типу. Множественные стигмы дизэмбриогенеза
На 12-е сутки жизни девочка переведена в отделение патологии новорожденных ГБУЗ РКБ им. Г.Г. Куватова, где находилась в течение месяца на долечивании и выхаживании. Заключительный клинический диагноз: основной — задержка внутриутробного развития по гипопластическому типу; сопутствующий — респираторный дистресс-синдром, осложненный пневмонией тяжелой степени, острое течение, дыхательная недостаточность 0-й степени, дыхательная недостаточность 3-й степени (ИВЛ) нивелирована (реконвалесцент). Ранняя анемия недоношенных тяжелой степени, купирована. Церебральная ишемия 1-й степени, острый период, синдром угнетения. Преретинопатия недоношенных. Функционирующий артериальный проток. Врожденная аномалия развития шейного отдела позвоночника. Гипоплазия шейных позвонков. Укорочение левых верхних и нижних конечностей (гемигипоплазия). Незрелость тазобедренных суставов. Задержка внутриутробного развития, врожденная гипотрофия 3-й степени. Постнатальная гипотрофия 1-й степени. Постконцептуальный возраст 40 нед.
В связи с текущей дезинфекцией родильного дома ребенок переведен для дальнейшего наблюдения и лечения в отделение патологии новорожденных ГБУЗ РДКБ г. Уфы, где находился 12 дней с основным диагнозом: недоношенность 34 нед и сопутствующим диагнозом: задержка внутриутробного развития. Гипотрофия 3-й степени. Перинатальное поражение ЦНС, острый период, синдром двигательных нарушений.
На 52-х сутках жизни состояние ребенка стабилизировалось, масса тела составила 1990 г, рост — 39 см; девочку с мамой выписали домой. Были даны рекомендации:
1) наблюдение участкового педиатра в группе здоровья 2Б; 2) свободное грудное вскармливание; 3) ежедневные прогулки от 15–30 мин до 4–5 ч, дробно, в теплую погоду;
4) ежедневные гигиенические ванны; 4) общие анализы крови и мочи ежемесячно; 5) медицинский отвод от профпрививок до 6 мес.; 5) эхоКГ в 6 мес.; 6) наблюдение невролога, окулиста, ортопеда по месту жительства; 7) витамин D (колекальциферол 15000 МЕ/мл) по 2 капли 1 р/день per os круглогодично; фолиевая кислота по 1/4 таблетки 2 р/день per os; эубиотики per os по инструкции.
В 5 мес. мама девочки обратилась в кабинет катамнеза, масса тела на момент обращения составила 2920 г, рост — 39,5 см, окружность груди — 31 см. Неврологический статус: ребенок активен, сознание ясное. Физическое развитие оценено как очень низкое, дисгармоничное. Гипотрофия 2-й степени. В связи с очень несимметричными пропорциями тела (большая голова относительно тела) предположили наличие гидроцефалии (рис. 1).
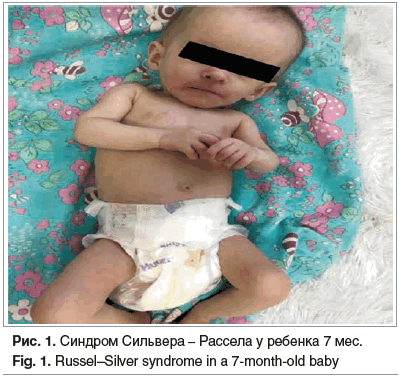
Девочку в экстренном порядке направили на нейросонографию, по результатам проведенного обследования патологии не выявлено. Заключение невролога: психомоторное развитие соответствует скорригированному возрасту, неврологический статус удовлетворительный. На консультации у гастроэнтеролога патология со стороны ЖКТ исключена, для кормления ребенка рекомендована смесь «Альфаре», прикорм — овощное пюре, каши, блюда коррекции, витамин D, массаж общеукрепляющий. В последующем девочка начала набирать вес. При втором посещении кабинета катамнеза в 6,5 мес.: физическое развитие очень низкое, масса тела 3172 г, рост 51 см, окружность головы 40 см, окружность груди 32 см. При осмотре определены характерные стигмы дизэмбриогенеза: карликовость, асимметрия рук, треугольные контуры лица с высоким лбом и мелкими чертами, маленький рот, узкие губы с опущенными углами, микрогнатия нижней челюсти, оттопыренные уши. Ярко выраженные лобные бугры черепа, мозговая часть черепа непропорционально велика по отношению к лицевой, что создает картину псевдогидроцефалии. Девочка активна, голову держит, сидит с поддержкой, следит за игрушками, контактна.
Результаты лабораторных и инструментальных исследований, проведенных в кабинете катамнеза
Клинический анализ крови и мочи: показатели в пределах нормы. Биохимическое исследование крови патологии не выявило. Копрограмма в норме.
Нейросонография: боковые желудочки не расширены. Сосудистые сплетения с четкими ровными контурами Мозговой кровоток при допплерометрии не изменен. Электроэнцефалография: нейрофизиологическая незрелость, ирритативные изменения ритма. Отоакустическая эмиссия: тест пройден ЭхоКГ: ультразвуковые признаки открытого овального окна (0,2 мм). ЭКГ: синусовая тахикардия, ЧСС 140 в минуту, ЭОС вертикальная, повышение электрических потенциалов межжелудочковой перегородки УЗИ органов брюшной полости, почек, тимуса, тазобедренных суставов: структурных изменений не выявлено.
Ребенок направлен в республиканский генетический центр, где выставлен диагноз ССР (возраст девочки на этот момент составил 7 мес.). Кровь пробанда и родителей исследована в ФГБНУ «МГНЦ», получено заключение: аномальное метилирование гена Н19 (определено методом микросателлитного анализа локуса 7-й хромосомы). Данное молекулярно-генетическое обследование позволило подтвердить наличие ССР и выставить диагноз: Q87.1 Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью. Синдром Сильвера — Рассела, аномальное метилирование гена Н19.
Заключение
Представленное клиническое наблюдение недоношенного ребенка первого года жизни демонстрирует нетяжелое течение ССР. Подозрение на генетическую патологию возникло лишь при втором посещении кабинета катамнеза, ближе к 7 мес жизни, в связи с отставанием в физическом развитии и имеющимися стигмами дизэмбриогенеза. Многие симптомы — признаки задержки внутриутробного развития, низкая прибавка массы тела и отставание в росте после рождения — могут быть проявлением других заболеваний, характерных для маловесных и недоношенных детей. Выявленные многочисленные стигмы дизэмбриогенеза должны мотивировать неонатолога и педиатра на проведение как можно более ранней консультации генетика с дальнейшим молекулярно-генетическим обследованием с целью своевременной диагностики наследственного заболевания.
Сведения об авторах:
Хафизова Наиля Римовна — к.м.н., доцент кафедры педиатрии с курсом ИДПО ФГБУ ВО БГМУ Минздрава России; 450008, Россия, г. Уфа, ул. Ленина, д. 3; ORCID iD 0000-0002-1452-9998.
Мерзлякова Динара Рафкатовна — аспирант кафедры педиатрии с курсом ИДПО ФГБУ ВО БГМУ Минздрава России; 450008, Россия, г. Уфа, ул. Ленина, д. 3; ORCID iD 0000-0001-9037-7124.
Сафина Юлия Фагилевна — клинический ординатор кафедры педиатрии с курсом ИДПО ФГБОУ ВО БГМУ Минздрава России; 450008, Россия, г. Уфа, ул. Ленина, д 3.
About the authors:
Nailya R. Khafizova — Cand. of Sci. (Med.), associate professor of the Department of Pediatrics with the Course of Additional Professional Education, Bashkir State Medical University; 3, Lenin str., Ufa, 450008, Russian Federation; ORCID iD 0000-0002-1452-9998.
Dinara R. Merzlyakova — post-graduate student of the Department of Pediatrics with the Course of Additional Professional Education, Bashkir State Medical University; 3, Lenin str., Ufa, 450008, Russian Federation; ORCID iD 0000-0001-9037-7124.
Yuliya F. Safina — clnical resident of the Department of Pediatrics with the Course of Additional Professional Education, Bashkir State Medical University; 3, Lenin str., Ufa, 450008, Russian Federation.
1. Андреева Л.П., Кулешов Н.П., Мутовин Г.Р. и др. Наследственные и врожденные болезни: вклад в детскую заболеваемость и инвалидность, подходы к профилактике. Педиатрия. 2007;3:8–14.
2. Коровкина Е.А., Жилина С.С., Конюхова М.Б. и др. Синдром Сильвера — Рассела: анализ клинического полиморфизма. Детская больница. 2008;3(33):14–18.
3. Новиков П.В. Семиотика наследственных болезней у детей (симптом — синдром — болезнь). М.: Триада-Х; 2009.
4. Bliek J., Terhal P., van den Bogaard M.J. et al. Hypomethylation of the h19 gene causes not only Silver — Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet. 2006;78:604–614.
5. McCann J.A., Zheng H., Islam A. et al. Evidence against GRB10 as the gene responsible for Silver — Russell syndrome. Biochem Biophys Res Commun. 2001;286:943–948.
6. Monk D., Bentley L., Hitchins M. et al. Chromosome 7p disruptions in Silver — Russell syndrome: delineating an imprinted candidate gene region. Hum Genet. 2002;111:376–387.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Синдром Сильвера-Рассела - клиника, диагностика
Изменения кожи при заболеваниях щитовидной железы (тиреотоксикозе)
Определение. Группа системных заболеваний, клинически проявляющихся поражением щитовидной железы с развитием синдрома тиреотоксикоза в сочетании с экстратиреоидной патологией и возможным появлением меланинового меланоза. Среди заболеваний с тиреотоксикозом чаще всего встречается диффузный токсический зоб (болезнь Грейвса) (на его долю приходится до 80% всех случаев тиреотоксикозов).
Историческая справка. Впервые диффузный токсический зоб был описан в 1825 г. Калебом Пари, в 1835 г.— Робертом Грейвсом, а в 1840 г. — Карлом фон Базедовым.
Этиология и патогенез меланоза кожи. Гиперпигментация кожи является результатом вторичного гипокортицизма. Он возникает вследствие повышенного метаболизма кортикостероидов, который приводит к развитию хронической надпочечниковой недостаточности.
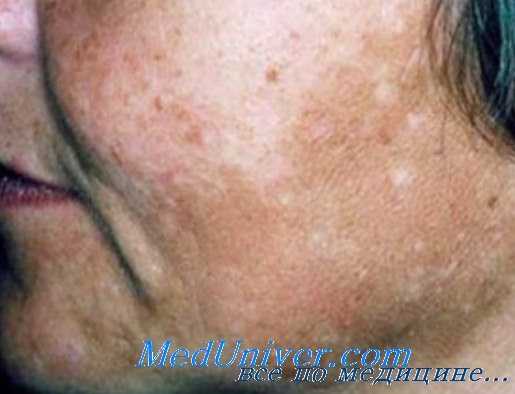
Мелазма при тиреотоксикозе
Частота возникновения меланоза. Гиперпигментация при тиреотоксикозах развивается в 10% случаев.
Изменения кожи и ее придатков. Кожный покров становится теплым и влажным. Сосуды кожи расширены (компенсаторная реакция для отдачи тепла), поэтому она теплая на ощупь и влажная. Ногтевые пластинки начинают слоиться, истончаются, становятся мягкими, склонны к повышенной ломкости.
Гиперпигментация кожи может быть диффузной, как при болезни Аддисона, но чаще протекает по типу мелазмы. Пигментация чаще проявляется в местах трения (шея, поясница, локоть и другие), а также в складках на ладонях и подошвах. У некоторых пациентов с болезнью Грейвса выявляется витилиго, крапивница, очаговая алопеция.
За счет высокой эластичности кожи у больных долго не образуются морщины, и они часто выглядят моложе своих лет. Подкожно-жировой слой развит слабо. Характерным признаком является выраженная пигментация век (симптом Jellinek). Stefan Jellinek (1871-1968) — австрийский врач, который первым описал гиперпигментацию верхних век при базедовой болезни.
Приблизительно у 5% пациентов с болезнью Грейвса встречается претибиальная микседема. Сначала плотные папулы или бляшки располагаются асимметрично на обеих ногах. Они растут, сливаются и в тяжелых случаях охватывают всю поверхность голеней и тыла стоп.
Дифференцируют с болезнью Аддисона. При тиреотоксикозах и болезни Аддисона пигмент может скапливаться в кожных складках ладоней и подошв. Однако при тиреотоксикозах слизистые оболочки, как правило, не пигментируются, а пигментация сосков и кожи гениталий менее выражена.
Некоторые авторы при тиреотоксикозах описывают гиперпигментации на нижних конечностях (преимущественно на голенях), спине и ногтевом ложе. При гистологическом исследовании пигментированной кожи у таких пациентов обнаруживается меланин в эпидермисе и отложения гемосидерина вокруг капилляров и потовых желез. Лечение мерказолилом вызывало незначительное ослабление пигментации.
Расположение очагов поражения, отложения гемосидерина и плохой ответ гиперпигментаций на лечение могут помочь в проведении дифференциальной диагностики таких тиреотоксикозов от болезни Аддисона.
Видео методики пальпации щитовидной железы
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Определение. Наследственное заболевание, основными признаками которого являются: маленький рост, треугольное лицо с небольшой нижней челюстью и нарушение симметрии всего тела.
Историческая справка. Впервые заболевание описали американский педиатр Н. К. Silver с соавторами в 1953 г. и английский педиатр A. Russell в 1954 г..
Этиология и патогенез. Встречается в большинстве случаев спорадически, однако описаны и единичные родословные с данной патологией. Предполагаются аутосомно-доминантный и аутосомно-рецессивный типы наследования.
Частота. В популяции 1:30000.
Возраст и пол. Болезнь проявляется уже в пренатальном (внутриутробном) периоде. Половых различий нет.
Клиника. Характерна пренатальная задержка роста. Дети рождаются небольшой длины (до 45 см) и с малой массой тела (1,5—2,5 кг). С годами отставание в росте сохраняется, в связи с чем окончательный рост у женщин менее 150 см, у мужчин немногим выше 150 см. Масса тела у взрослых нормальная или даже избыточная.
Часты изменения мочеполовой системы: крипторхизм, гипоспадия, гипоплазия полового члена, мошонки, аномалии почек. Характерна асимметрия тела (лица, туловища, длины ног). Лицо треугольной формы: псевдогидроцефалия, большой лоб и гипоплазия нижней челюсти, высокое нёбо, нередко с расщелиной, оттопыренные уши.
Клиндактилия V пальцев за счет девиации дистальной фаланги, узкая грудная клетка, короткие руки, поясничный лордоз. Описаны случаи сочетания карциномы печени и данного синдрома. Интеллект обычно нормальный.
Поражения кожи. Имеются кофейные пятна округлой формы, размерами от 1 до 30 см.
Диагноз. Основными клиническими проявлениями считаются: пренатальная задержка роста, асимметрия тела, значительное отставание массы тела от нормы, клиндактилия V пальца, псевдогидроцефалия, треугольное лицо, крипторхизм.
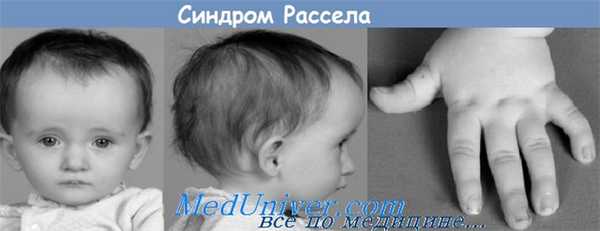
Заболевания, ассоциированные с пятнами по типу «кофе с молоком»
| Заболевание | Другие поражения кожи | Системные поражения |
| Нейрофиброматоз | Веснушки в подмышечной области, узелки Лиша (радужка), нейрофибромы | Аномалии скелета, неврологические поражения |
| Синдром Олбрайта | Нет, только небольшое количество распространенных пятен по типу «кофе с молоком» | Преждевременное половое созревание у девочек, полиостозная фиброзная дисплазия |
| Синдром Уотсона | Веснушки в подмышечной области | Стеноз легких, задержка психического развития |
| Карликовость Сильвера-Рассела | Гипогидроз в грудном возрасте | Карликовость, асимметричность скелета, клинодактилия V пальца руки |
| Атаксия-телеангиэктазия | Телеангиэктазия на бульварных конъюнктивах и на лице, изменения по типу склеродермии | Задержка роста, атаксия, задержка психического развития, лимфопения, IgA, IgE, инфекции верхних дыхательных путей |
| Туберозный склероз | Гипопигментированные пятна, шагреневая бляшка, аденома сальных желез, подногтевые фибромы | Центральная нервная система, почки, сердце, легкие |
| Синдром Тернера | Дряблая кожа, особенно вокруг шеи; лимфедема в грудном возрасте, гемангиомы | Карликовость, дисгенезия гонад, аномалии скелета, аномалии почек, дефекты сердечно-сосудистой системы |
| Синдром Блума | Телеангиэктатическая эритема на щеках, фоточувствительность, ихтиоз | Низкорослость, малярная гипоплазия скуловых костей, риск развития злокачественных опухолей |
| Множественные лентиго (LEOPARD-синдром) | Лентиго, веснушки в подмышечной области | Отклонения в показателях электрокардиограммы, глазной гипертелоризм, стеноз легких, аномалии развития гениталий, задержка роста, нейросенсорная глухота |
| Синдром Вестерхофа | Гипопигментированные пятна | Задержка роста и психического развития |
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром Рассела-Сильвера
Синдром Рассела–Сильвера – это редкое генетическое нарушение, характеризующееся задержкой внутриутробного развития и постнатальной низкорослостью. Другими фенотипическими маркерами синдрома являются макроцефалия, лицо треугольной формы, нарушение пропорций тела, клинодактилия, пигментные пятна на коже. На диагностическом этапе учитываются клинические симптомы, проводится исследование эндокринной функции, определение костного возраста, генетические исследования. Лечение ограничено симптоматическими мерами (гормонотерапия, корригирующие операции, физиотерапия).
МКБ-10
Общие сведения
Заболевание названо в честь врачей-педиатров, описавших его практически одновременно, но независимо друг от друга: Г. Сильвера (1953 г.) и А. Рассела (1954 г.). Синдром встречается с частотой 1 случай на 30-100 000 новорожденных. Половых и национальных различий в распространенности не отмечено. Патологию отличает клиническая неоднородность и широкий спектр проявлений, затрагивающих практически все системы органов: сердечно-сосудистую, эндокринную, костно-мышечную, мочеполовую, нервную и др. Ведение детей с синдром Рассела–Сильвера требует квалифицированного междисциплинарного подхода.
Причины
Генетические аномалии, лежащие в основе синдрома Рассела–Сильвера, различны – именно этот фактор обусловливает гетерогенность проявлений и тяжести патологии. Наиболее распространенными генетическими вариациями являются гипометилирование генов на хромосоме 11p15 (наблюдается у 30-60% пациентов) и однородительская материнская дисомия по 7-й хромосоме (встречается у 5-10% пациентов). Тем не менее, примерно у 40% детей молекулярно-генетическая основа синдрома Сильвера–Рассела остается невыясненной.
Как правило, генетические дефекты возникают случайно, однако известны отдельные случаи наследственной передачи патологических генов. Случайные мутации обусловлены патологией беременности или неблагоприятными внешними воздействиями в ранние периоды эмбриогенеза.
Патогенез
В настоящее время считается, что развитие синдрома Рассела–Сильвера обусловлено нарушением геномного импринтинга – активности генов в зависимости от их материнского или отцовского происхождения. И в 7, и в 11 хромосоме располагаются кластеры импринтируемых генов. Часть детей с данной патологией имеет мультилокусное нарушение импринтинга в нескольких областях или на других хромосомах.
В области хромосомы 11p15. 5. находятся два центра импринтинга − ICR1 и ICR2. Центр ICR1 обеспечивает активность генов, играющих важнейшую роль в регуляции роста плода. В частности, это материнский ген H19 (ген скелетной мускулатуры) и отцовский ‒ IGF2 (инсулиноподобный фактор роста 2, ИФР2). Гипометилирование приводит к их инактивации и задержке роста. Известно, что генетические абберации данного локуса также связаны с развитием синдрома Беквита-Видемана, напротив, характеризующегося макросомией.
Около 10% людей с синдромом Сильвера–Рассела имеют обе копии материнской 7 хромосомы, а не по одной копии от каждого родителя. Это называется однородительской материнской дисомией. На 7-ой хромосоме в сегменте 7р11.2 расположены импринтированные гены IGFBP1 (белок, связывающий ИФР1), IGFR (рецептор ИФР) и GRB10 (белок, связывающий гормон роста 10), которые также ответственны за рост и развитие организма. Точный механизм, посредством которого эти гены вызывают нарушения роста, до конца не изучены.
Классификация
В литературе и научных публикациях упоминается о двух клинических формах синдрома Рассела–Сильвера: «тяжелой» и «мягкой». Считается, что дефекты седьмой хромосомы в большинстве случаев связаны с более легким течением синдрома. Это выражается в отсутствии серьезных аномалий развития внутренних органов и не столь выраженном дефиците соматотропного гормона.
Симптомы
Физическое и психическое развитие
Дети с синдромом Рассела–Сильвера рождаются маловесными (менее 2500 г), небольшого роста (менее 45 см). Примерно треть из них появляется на свет недоношенными, однако даже в этих случаях отмечается несоответствие антропометрических показателей сроку гестации. Данное состояние расценивается как примордиальный нанизм. Дети отличаются пониженным аппетитом, плохо набирают вес.
Телосложение больных чаще асимметричное по типу гемигипоплазии. Дефицит роста сохраняется на протяжении всей жизни. В зарубежных исследованиях сообщается, что рост взрослых с синдромом Сильвера–Рассела, не получавших лечения гормоном роста, составляет примерно 150 см у мужчин и 140 см у женщин.
Фенотипические особенности
Больные синдромом Рассела–Сильвера отличаются наличием ряда характерных стигм дизэмбриогенеза. В их числе относительная макроцефалия с выступающими лобными буграми, лицо в форме треугольника, лопоухость, готическое небо и др. Часто имеются пороки зубо-челюстной системы: недоразвитие нижней челюсти, микродентия, скученность зубов. У детей маленький рот с тонкими губами с опущенными вниз уголками («рот карпа»). На коже определяются пигментные пятна типа «кофе с молоком».
Скелетные аномалии
У пациентов имеются множественные отклонения в развитии костной системы. Типично позднее зарастание родничков. Рентгенологически определяются добавочные пястные кости, остеопороз, задержка костного возраста по сравнению с паспортным. Возможен врожденный вывих бедра, плоскостопие. Характерными признаками синдрома Рассела-Сильвера служат клинодактилия мизинцев кистей, синдактилия 1-го и 2-го пальцев стоп. Грудная клетка узкая, воронкообразная, трубчатые кости конечностей укорочены, имеется поясничный гиперлордоз.
Аномалии внутренних органов
Синдром ассоциирован с различными патологиями внутренних органов. Кардиальные нарушения чаще всего представлены синусовой аритмией, блокадой ножек пучка Гиса, сердечными пороками (пролапсом митрального или трикуспидального клапана, дополнительной хордой желудочка). Большой вариативностью и степенью тяжести отличаются сопутствующие патологии ЖКТ – от гастроэзофагеального рефлюкса и дискинезии ЖВП до гепатоцеллюлярной карциномы.
Наиболее частыми аномалиями мочеполовой системы при синдроме Сильвера–Рассела являются недоразвитие гениталий, гипоспадия, крипторхизм, аплазия матки. Ренальные синдромы включают почечный канальцевый ацидоз, метаболическую нефропатию и др.
Нервно-психическое развитие
В младенчестве и раннем детстве пациенты могут иметь задержку моторного развития из-за выраженной мышечной гипотонии, асимметрии конечностей. Также нередко встречается отставание развития речи, особенно у пациентов с дефектом 7 хромосомы, перинатальным поражением ЦНС. У большинства больных интеллект близок к норме, однако встречаются случаи, протекающие с ЗПР, поведенческими нарушениями, расстройствами аутистического спектра.
Эндокринная дисфункция
Гормональный профиль пациентов показывает наличие у них дефицита соматотропного гормона, инсулиноподобного фактора роста 1, гиперпролактинемии. Примерно в 30% случаев отмечено преждевременное половое созревание: ранние менструации у девочек, оволосение и мутация голоса – у мальчиков. Младенцы и дети подвержены развитию ночных эпизодов гипогликемии. Однако во взрослом возрасте увеличивается риск метаболических расстройств и нарушения толерантности к глюкозе.
Осложнения
Множественные структурные патологии органов и функциональные расстройства у лиц с синдромом Рассела–Сильвера обусловливают риск развития различных осложнений. Асимметрия тела и ортопедические проблемы связаны с повышенным травматизмом вследствие сложностей с удержанием равновесия и ходьбой. Сердечно-сосудистые и почечные аномалии могут вызвать недостаточность работы органов (ХСН, ХПН). Из-за метаболических нарушений пациенты старшего возраста склонны к набору лишнего веса, повышенному кровяному давлению, гиперлипидемии. Отмечена корреляция синдрома с опухолевыми процессами (рак печени, рак яичка, аденома гипофиза).
Диагностика
Клинические критерии, необходимые для установления диагноза синдрома Рассела–Сильвера, включают микросомию при рождении, характерные фенотипические признаки (диспропорции тела, треугольное лицо, клинодактилия и др.). При первичном обращении проводится антропометрия, оценка роста и развития ребенка. Для подтверждения диагностической гипотезы требуется консультация генетика, а также лабораторно-инструментальное дообследование:
- Рентгенография.Рентген кистей рук позволяет выявить несоответствие биологического возраста с паспортному. Другие исследования скелета необходимы для обнаружения аномалий опорно-двигательного аппарата: выполняется Rg черепа, челюстей, позвоночника, грудной клетки, стоп, суставов. Функциональное состояние органов оценивается с помощью рентгенографии желудка, экскреторной урографии.
- Лабораторная диагностика. Назначается анализ гормонального профиля: соматотропина, соматомедина С, пролактина, гонадотропинов, ТТГ. С целью определения степени дефицита СТГ выполняются стимуляционные фармакологические пробы. При склонности к гипогликемии осуществляется суточный мониторинг глюкозы.
- Генетические исследования. Определяется кариотип пациентов (при синдроме Рассела-Сильвера выявляется нормальный женский или мужской кариотип). Ключевым моментом в установлении диагноза служит молекулярно-генетический анализ, позволяющий выявить хромосомные нарушения.
- Исследование внутренних органов. Скрининговым методом выявления пороков внутренних органов служит сонография. Наиболее информативными являются УЗИ брюшной полости и почек, УЗИ мошонки, УЗИ матки и придатков, ЭхоКГ. Для установления причин соматотропной недостаточности показана МРТ гипофиза. Нарушения сердечного ритма фиксируются при проведении ЭКГ или ХМ ЭКГ.
Дифференциальная диагностика
Новорожденных с синдромом Рассела-Сильвера, необходимо отличать от детей с маловесностью и низкорослостью, вызванных другими причинами:
- идиопатической ЗВУР;
- синдромом Блума;
- синдром Ниймеген;
- 3М-синдромом;
- нанизмом Mulibrey;
- неонатальной прогерией.
Лечение синдрома Рассела-Сильвера
Радикальное излечение и полное выздоровление невозможно. Терапия носит поддерживающий характер, направлена на улучшение внешнего вида, предупреждение осложнений и повышение качества жизни. Пациентов с синдромом Сильвера-Рассела наблюдают врачи-эндокринологи, ортопеды, урологи, педиатры. Основные направления лечения включают:
- Нутритивную поддержку. В первые годы жизни важнейшей задачей является обеспечение нормального питания ребенка. Это необходимо как для адекватного роста и набора массы тела, так и для предупреждения гипогликемии. Если ребенок отказывается от еды, используется парентеральное питание, кормление через назогастральный зонд.
- Заместительную гормонотерапию. С целью оптимизации роста, увеличения мышечной массы, улучшения двигательной функции назначается терапия рекомбинантным гормоном роста человека (РГРЧ). Для нормального полового развития применяется лечение гонадостероидами (тестостерон, синестрол, ХГЧ). При дефиците ТТГ используются тиреоидные гормоны.
- Симптоматическое лечение. Пациентам с ГЭРБ назначается терапия Н2-блокаторами или ингибиторами протонной помпы. Возможно проведение хирургического лечения – фундопликации. Пациентам с ортопедическими проблемами показано ношение лечебной обуви, стелек. Стоматологические проблемы могут потребовать ортодонтического лечения. Мальчикам необходима хирургическая коррекция гипоспадии, крипторхизма.
- Реабилитационные мероприятия. Улучшить двигательные функции, стимулировать физическое развитие помогает общий массаж, физиопроцедуры (электрофорез, парафиновые аппликации), лечебная физкультура, гидрокинезиотерапия.
Прогноз и профилактика
Большинство взрослых людей с синдромом Рассела-Сильвера, получающих необходимое лечение, имеют приемлемое качество жизни и нормальную фертильность. Значительная часть аномалий поддается коррекции, что позволяет пациентам вести полноценную жизнь. Однако рост таких больных все равно отстает от средних значений в популяции.
Профилактика тесно связана с антенатальными мероприятиями, в первую очередь – предупреждением осложнений беременности. Заподозрить синдром Рассела-Сильвера у плода позволяет пренатальная ультразвуковая и генетическая диагностика. В этом случае родителей предупреждают о возможных отклонениях в физическом развитии ребенка, однако синдром не является абсолютным показанием к досрочному прерыванию беременности. Вероятность рождения второго ребенка с такой же патологией в одной семье крайне низкая.
1. Cиндром Сильвера-Рассела у ребенка двух лет: клинический случай из практики/ Крючкова Т.А., Мезенцева О.А.// Научные ведомости Белгородского государственного университета. Серия: Медицина. Фармация. – 2016.
2. Синдром Сильвера-Рассела: клиникогенетический анализ/ Коровкина Е.А., Жилина С.С., Конюхова М.Б.// Педиатрия. Журнал им. Г.Н. Сперанского. – 2011.
3. Случай дилатационной кардиомиопатии у девочки 3 лет с синдромом Сильвера-Рассела/ Лебеденко А.А., Сарычев А.М., Тарасова Е.А. и др.// Медицинский вестник юга России. – 2012.
4. Diagnosis and management of Silver–Russell syndrome: first international consensus statement/ E. L. Wakeling, F. Brioude, I. Netchine// Nature Reviews Endocrinologyvolume. - 13 (2017).
Синдром MERRF ( Миоклонус-эпилепсия с рваными красными волокнами )
Синдром MERRF – это редкое генетическое заболевание, которое вызвано структурными и биохимическими дефектами митохондрий, характеризуется тяжелым поражением центральной нервной системы и мышечной ткани. Клиническая картина может отличаться даже внутри одной семьи. Симптомы включают различные виды эпилептических припадков, нарушение координации, мышечную слабость. Подтверждающими диагностическими методами являются гистологическое исследование мышечного биоптата и ДНК-анализ. Специфического лечения не существует, проводится симптоматическая терапия нейрометаболическими, противосудорожными средствами, препаратами, улучшающими митохондриальную функцию.
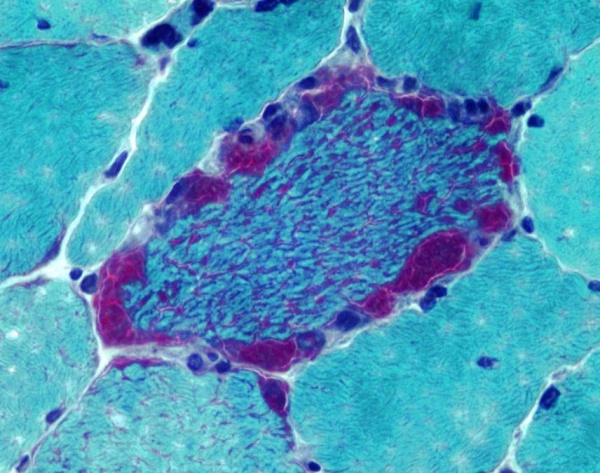
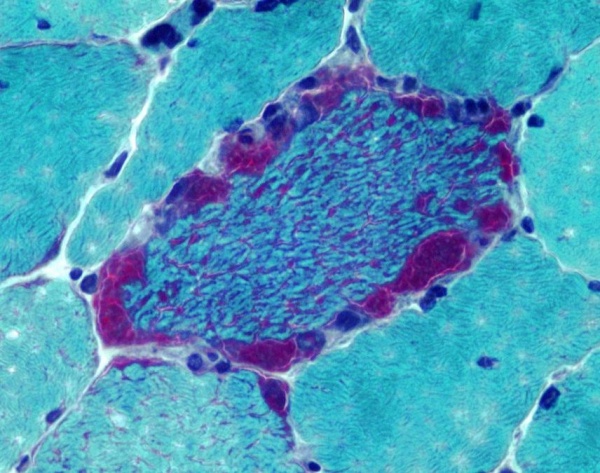
Синдром MERRF относится к группе наследственных митохондриальных энцефаломиопатий. MERRF представляет собой аббревиатуру, означающую «миоклонус (М) эпилепсия (Е) с рваными (R) красными (R) волокнами (F)». Заболевание впервые было описано японским неврологом Н. Фукухара в 1980 году. По различным эпидемиологическим данным, распространенность синдрома MERRF составляет от 0,25:100000 до 1,5:100000 населения. Наследование происходит только по материнской линии, при этом оба пола страдают с одинаковой частотой.
Возникновение синдрома MERRF вызвано точечными мутациями митохондриальной ДНК, а именно, изменениями нуклеотидной последовательности. Наиболее частыми считаются замена аденина на гуанин в положении 8344 транспортной РНК лизина или замена тимина на цитозин в положении 8356. Это приводит к нарушению образования митохондриальных белков.
Клиническое развитие синдрома MERRF зависит от количества дефектных ДНК, т.е. чем больше митохондриальных ДНК, имеющих мутацию, тем выше риск заболевания. Также возможно абсолютно бессимптомное носительство мутантных генов. Какие-либо значимые факторы риска, способные спровоцировать манифестацию данной патологии, отсутствуют.
В результате генетической мутации в митохондриях происходит сбой синтеза ферментов дыхательной цепи (цитохромоксидазы, сукцинатдегидрогеназы и др). Нарушаются процессы окислительного фосфорилирования – наиболее важного этапа в энергетическом обмене клеток, который обеспечивает образование основного количества молекул АТФ (универсального источника энергии для клеток).
Данные явления протекают практически во всех клетках организма. Наиболее сильно поражающий эффект сказывается на органах с высокой энергетической потребностью – центральной нервной системе и мышцах. Вследствие тканевой гипоксии в мышцах накапливается избыток молочной кислоты, что приводит к повреждению мышечных волокон.
В современной неврологии четких официальных разделений синдрома MERRF на отдельные формы не принято. В зависимости от количества точечных мутаций и клинических проявлений можно выделить следующие разновидности:
- Бессимптомная – имеется генетическая мутация с возможностью передачи по наследству при полном отсутствии симптомов.
- Латентная – «мягкое течение» без вовлечения нервной системы. Пациентов может беспокоить умеренная мышечная слабость.
- Манифестная – яркая клиническая картина с тяжелым течением и неблагоприятным прогнозом.
Синдром MERRF может дебютировать практически в любом возрасте. Начало в детском возрасте ассоциировано с более неблагоприятным прогнозом. Ранними признаками считаются повышенная мышечная утомляемость, ухудшение переносимости физических нагрузок, ноющие боли в икроножных мышцах. Из-за постепенного прогрессирования миопатии мышечная слабость приобретает более выраженный характер – пациент испытывает трудности при подъеме по лестнице, ходьбе. В тяжелых случаях больной с большим усилием может подняться с постели.
Типичны эпилептические припадки, которые могут быть разнообразными – непроизвольные подергивания мышц лица или рук без потери сознания (миоклонии), пароксизмы по типу кивков головой, генерализованные тонико-клонические припадки с потерей сознания. Миоклонии часто провоцируются резкими звуками или вспышкой света. Нарушается координация, возникает неустойчивость при ходьбе, стоянии.
Существенно страдают когнитивные функции, снижается память, концентрация внимания. Постепенно ухудшается зрение и слух. В ряде случаев наблюдаются периферические невропатии, проявляющиеся онемением, ощущением жжения, покалывания (парестезиями) в конечностях. Очень редко встречаются кожные образования – липомы, гемангиомы, бородавчатые невусы.
Синдром MERRF считается тяжелым заболеванием с большим числом осложнений. Наиболее неблагоприятными из них являются эпилептический статус и отек головного мозга. Вследствие тяжелой миопатии мышц глотки возможно нарушение глотания и попадание пищи в дыхательные пути, что приведет к развитию аспирационной пневмонии. Также из-за слабости дыхательных мышц возникает дыхательная недостаточность.
Атрофия зрительных нервов и пигментная дегенерация сетчатки у части пациентов вызывает полную потерю зрения. При ранней клинической манифестации ребенок значительно отстает в физическом и нервно-психическом развитии. Нарушение равновесия повышает риск падений и переломов. Очень редко наблюдается хронический панкреатит и сахарный диабет.
Пациентов с синдромом MERRF курируют врачи-неврологи. Больные детского возраста находятся под совместным наблюдением детских невропатологов и педиатров. При осмотре обращается внимание на общее снижение мышечного тонуса, ослабление сухожильных и наличие патологических рефлексов (Бабинского, Оппенгейма), невыполнение координационных тестов – позы Ромберга, пяточно-коленной, пальце-носовой проб. Дополнительное обследование включает:
- Лабораторные исследования. В биохимическом анализе крови отмечается увеличение концентрации молочной кислоты. В ликворе выявляется высокое содержание белка.
- ЭЭГ. На электроэнцефалограмме удается обнаружить эпилептиформную активность – генерализованные комплексы спайк-волна, диффузные медленные волны.
- Томография. На МРТ головного мозга видна атрофия коркового и белого вещества больших полушарий и мозжечка, кальцификация базальных ганглиев.
- ЭНМГ. При проведении электронейромиографии отмечается снижение амплитуды и длительности потенциалов двигательных единиц, что свидетельствует о поражении мышечной ткани.
- Гистологическое исследование. Один из главных диагностических тестов синдрома MERRF. В мышечном биоптате выявляются признаки атрофии – уменьшение размеров мышечных волокон, их бледное окрашивание, склероз эндомизия и перимизия. При окраске гистологических срезов по методу Гомори более чем в 5% мышечных волокон обнаруживается наличие «рваных красных волокон».
- ДНК-анализ. Основной метод для верификации диагноза. При молекулярно-генетическом исследовании находят мутации в митохондриальной ДНК – A8344G или Т8356С.
Синдром MERRF следует дифференцировать с другими митохондриальными заболеваниями (MELAS-синдром), а также с наследственными метаболическими расстройствами, поражающими мышечные ткани и нервную систему:
- миоклонус-эпилепсией Унферрихта-Лундборга;
- болезнью Лафоры;
- болезнями накопления (болезнь Гоше, лейкодистрофии, ганглиозидозы).
Лечение синдрома MERRF
На сегодняшний день эффективных способов терапии данной патологии не существует. Все мероприятия носят симптоматический и паллиативный характер, направлены на улучшение состояния пациента. Для замедления атрофических процессов в мышечной ткани обязательны занятия лечебной физкультурой. Регрессу симптоматики способствует диета с ограничением углеводов.
Детям с умственной отсталостью рекомендуются индивидуальные консультации дефектолога, психолога, логопеда. Лекарственная терапия проводится по общим принципам лечения митохондриальных болезней и включает 2 основные группы медикаментов:
- Метаболические препараты. С целью улучшения процессов клеточного дыхания применяется комплекс из энерготропных препаратов («митохондриальный коктейль»), в который входят кофакторы ферментов (витамины группы В), средства, стимулирующие перенос электронов в дыхательной цепи (коэнзим Q10) и антиоксидантов (аскорбиновая кислота, токоферол).
- Антиконвульсанты. Для предупреждения эпилептических припадков назначаются противосудорожные лекарственные средства – клоназепам, ламотриджин, топирамат. Вальпроевая кислота и ее производные строго противопоказаны, так как они угнетают митохондриальную функцию. Их применение приводит к резкому ухудшению состояния больного.
Продолжительность жизни, вероятность летального исхода при синдроме MERRF может сильно варьировать у разных пациентов. При малосимптомном течении (отсутствие поражения нервной системы, умеренная миопатия), систематическом проведении нелекарственного и лекарственного лечения продолжительность жизни может не отличаться от таковой в общей популяции.
При развернутом течении прогноз крайне неблагоприятный. Летальный исход наступает в течение 10-15 лет от начала заболевания. Основными причинами смерти выступают эпилептический статус, аспирация, дыхательная недостаточность. Единственным специфическим методом профилактики является пренатальная диагностика и прерывание беременности.
1. Наследственные болезни нервной системы: Рук. для врачей/ Под ред. Ю.Е. Вельтищева, П.А. Темина. - 1998.
4. Молекулярно генетическая характеристика болезней дыхательной цепи митохондрий у детей: Автореферат диссертации/ Цыганкова П.Г. - 2012.
Читайте также: