Синдром Смита-Лемли-Опитца - этиология, клиника, диагностика
Добавил пользователь Евгений Кузнецов Обновлено: 08.01.2026
Синдром Смита-Лемли-Опитца - этиология, клиника, диагностика
Синдром Смита-Лемли-Опитца недавно стал объектом пристального внимания неврологов и психиатров из-за отчетливой взаимосвязи с аутистическими проявлениями. Возможно, данное заболевание имеет генетическую природу и тесно связано с аутистическими симптомами (см. ниже).
Синдром Смита-Лемли-Опитца (ССЛО) регистрируется с частотой около 1 на 70000 новорожденных. Характерна высокая перинатальная смертность и высокая частота мертворождений. Среди выживших детей не менее чем у 80% отмечаются тяжелые отклонения, а у оставшихся 20% изменения фенотипа выражены умеренно.
Синдром Смита-Лемли-Опитца (ССЛО) до сих пор диагностируется на основании типичных клинических признаков. Заболевание вызвано врожденным нарушением постскваленового синтеза холестерина. Нарушение синтеза холестерина при ССЛО вызвано наследственной мутацией гена 3-b-гидроксистерол-d-7 редуктазы (DHCR7). Повреждение гена DHCR7 приводит к нарушению синтеза холестерина и десмостерола, приводящее к повышению уровня 7DHC/8DHC, характерному снижению уровня холестерина и, что особенно важно, дисморфическому развитию.
Открытие синдрома Смита-Лемли-Опитца вызвало новые вопросы о роли путей биосинтеза холестерина в развитии человека. В настоящее время у 250 пациентов с синдромом Смита-Лемли-Опитца идентифицирована 121 мутация; разнообразие мутаций обеспечивает различную клиническую выраженность. Для воспроизведения некоторых аномалий развития, характерных для ССЛО, и выяснения патогенеза заболевания, было создано две генетических модели синдрома на мышах.
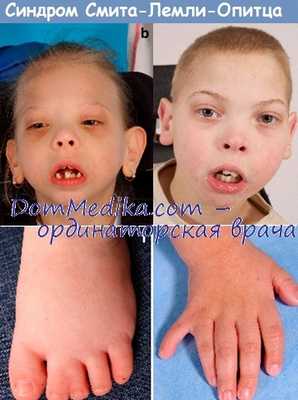
Наиболее характерными проявлениями синдрома наряду с задержкой умственного развития, аутизмом, другими поведенческими нарушениями и гипотонией является необычное строение лица с высоким плоским лбом, вывернутыми вперед ноздрями и микрогнатией, а также аномалии гениталий у мальчиков, включая крипторхизм и (или) гипоспадию, а в крайних случаях—неразвитые мужские гениталии, несмотря на нормальный кариотип XY (Scarbrough et al., 1986). Другие проявления включают аномальное строение ушных раковин, птоз век, эпикант, поперечное возвышение ладони и различные аномалии конечностей и внутренних органов (Opitz et al., 1994).
У некоторых пациентов отмечаются припадки, достаточно часто встречается отсутствие аппетита. В течение первого года жизни смертность высокая. Аномалии головного мозга могут включать микроцефалию, гипоплазию лобных долей, аномальное строение извилин и гипоплазию мозжечка. Случаи наиболее выраженного поражения иногда выделяются как ССЛО 2 типа (Curry et al., 1987).
Синдром наследуется аутосомно-рецессивным путем. Явное повышение частоты заболевания у мужчин, вероятно, связано с более легкой диагностикой на основании очевидных аномалий половых органов.
Концентрация холестерина в плазме очень низкая, в то время как содержание 7-гидрохолестерола повышено, что предполагает дефект фермента, приводящего к разрыву двойной связи гидрохолестерола (Tint et al., 1994). Выживаемость строго коррелирует с более высоким уровнем холестерина плазмы (Tint et al., 1995). Возможна пренатальная диагностика путем определения содержания 7-дегидрохолестерола в амниотической жидкости (Dallaire et al., 1995). Потребление большого количества холестерина может нормализовать его уровень в крови, но необязательно будет клинически эффективно (Ullrich et al., 1996).
Синдром Смита-Лемли-Опица
Синдром Смита-Лемли-Опица — это редкое наследственное заболевание, которое связано с генетическим дефектом обмена холестерина. Возникает при мутации гена DHCR7 в 11-й хромосоме, в локусе 11q12-q13. Патология проявляется низкорослостью, умственной отсталостью, многочисленными костными аномалиями и врожденными пороками внутренних органов. Диагностика синдрома предполагает биохимические анализы, генетическое тестирование, инструментальные способы визуализации соответственно превалирующей симптоматике. Лечение поддерживающее: нейрореабилитационные мероприятия, помощь пластических хирургов, кардиохирургов, челюстно-лицевых хирургов.
МКБ-10
Общие сведения
Синдром назван в честь американских педиатров Д. Смита, Л. Лемли и генетика Дж. Опица, которые в 1964 г. описали его клинические признаки. Генетические механизмы развития заболевания были установлены в 1998 г. Частота встречаемости синдрома составляет в среднем 1:29000, чаще болеют европейцы (1:22000). При этом носительство мутантного гена широко распространено: до 1,2% у светлокожего населения и до 0,8% у темнокожих людей. Состояние характеризуется тяжелым течением, частой инвалидизацией, высокой смертностью, что обуславливает его актуальность в современной генетике.
Причины
Синдром Смита-Лемли-Опица связан с аномалией (миссенс- и нонсенс-мутацией) гена DHCR7, который располагается на длинном плече 11 хромосомы (11q12-q13). Он кодирует фермент 7-дегидрохолестерин-редуктазу, участвующий в заключительном этапе синтеза холестерина. Заболевание имеет аутосомно-рецессивный тип наследования. Риск возникновения синдрома у ребенка, оба родителя которого являются носителями поврежденного гена, составляет 25%.
Патогенез
Основным механизмом, который провоцирует появление патогномоничных фенотипических признаков синдрома, считается холестериновый дефицит. При этом заболевании конечная молекула не образуется, вместо нее в организме накапливаются липопротеины низкой плотности, другие промежуточные метаболиты. Эти вещества в избыточных количествах обладают токсическим влиянием на клетки.
Неврологические нарушения вызваны недостаточным образованием липидов, которые необходимы для миелинизации нервных волокон, а специфический сигнальный белок sonic hedgehog провоцирует нарушения внутриутробного формирования мозговой ткани. Дефицит холестерина коррелирует с недостатком гормонов (тестостерона, эстрогенов), что объясняет нарушения формирования наружных половых органов у мальчиков.
Симптомы
Для болезни Смита-Лемли-Опица специфичны микроцефалия, волчья пасть, микрогнатия. Реже встречается синдром Пьера-Робена в сочетании с выступающими резцами, что создает затруднения при жевании пищи, проведении анестезиологического пособия. У 95% детей наблюдается Y-образная синдактилия 2-3 пальцев ног, в половине случаев отмечается постаксиальная синдактилия. Также характерны вывих тазобедренных суставов, укорочение конечностей.
При генетическом дефекте DHCR7 типично отставание в росте, физическом развитии. С самого рождения младенцы плохо набирают вес, длина их тела ниже нормы. По мере взросления ребенка задержка роста становится более заметной, во взрослом возрасте параметры тела находятся ниже 3-го перцентиля. Нарушается половое созревание: у мальчиков выявляются гипоспадия, крипторхизм, у девочек — гипертрофия клитора.
У большинства пациентов определяется выраженная неврологическая симптоматика. Она проявляется нарушениями сна (70% больных): частыми ночными пробуждениями, неспособностью уснуть, отсутствием четкого режима дня. Психические симптомы включают повышенную агрессивность, в том числе аутоагрессию, склонность к навязчивому поведению, расстройства аутистического спектра. Большинство детей, страдающих синдромом Смита-Лемли-Опица, умственно отсталые.
Осложнения
36-38% случаев синдрома осложняется врожденными сердечными пороками. Аномалии желудочно-кишечного тракта встречаются у 25-29% больных. Они включают гастроэзофагеальный рефлюкс, болезнь Гиршпрунга, мальротацию кишечника. Распространены пороки мочеполовой системы: кистозная дисплазия почек (29%), дистопия почек (19%), гидронефроз (16%) и обструкция лоханочно-мочеточникового соединения (13%). Около 12-18% пациентов страдают от снижения зрения.
Большинству больных устанавливается инвалидность из-за выраженной умственной отсталости, отставания темпов физического развития. Синдром Смита-Лемли-Опица ассоциирован с высоким уровнем смертности как в перинатальном периоде, так и после рождения ребенка вследствие грубых врожденных аномалий. Хотя многие пациенты доживают до зрелого возраста, им требуется постоянный уход и медицинское наблюдение.
Диагностика
При физикальном осмотре ребенка удается выявить патогномоничные изменения фенотипа, что вместе с признаками задержки роста и психомоторного развития позволяет поставить предварительный диагноз. Для подтверждения синдрома Смита-Лемли-Опица назначается обследование у генетика, ряд диагностических мероприятий:
- Биохимический анализ. В плазме крови и тканях организма проверяется содержание липидов. При этом генетическом заболевании наблюдается повышение уровня 7-дегидорохолестерина одновременно со снижением количества холестерина.
- Генетическое исследование. Для обнаружения специфической мутации выполняется секвенирование экзона. При выявлении аномалии DHCR7 диагноз синдрома Смита-Лемли-Опица подтверждается со 100% точностью. Такая же диагностика показана родственникам больного.
- Пренатальная диагностика. Парам с отягощенной наследственностью рекомендуется исследование во время беременности путем измерения концентрации 7-дегидрохолестерола, генетического анализа амниотической жидкости или ворсин хориона.
- Инструментальные методы. Пациентам проводится комплексное обследование сердечно-сосудистой (эхокардиография, ЭКГ), мочеполовой (экскреторная урография, УЗИ органов малого таза), нервной системы (ЭЭГ, нейросонография, КТ или МРТ головного мозга).
Лечение синдрома Смита-Лемли-Опица
Консервативная терапия
При наличии синдрома Смита-Лемли-Опица проводится симптоматическая терапия, которая подбирается индивидуально на основании ведущих клинических признаков. Пациентам требуется помощь мультидисциплинарной команды врачей, поскольку у одного больного сочетаются разнообразные поражения внутренних органов, нарушения костно-мышечной системы, нервно-психические расстройства. Выделяют следующие направления лечения генетического синдрома:
- Диетотерапия. Для нормализации липидного профиля плазмы крови применяются пищевые добавки с кристаллическим холестерином, рацион обогащается яичными желтками, животными жирами.
- Реабилитация. Для улучшения моторных функций назначается комплексная программа ЛФК, массаж, кинезитерапия. Чтобы скорректировать нервно-психический дефицит, показаны длительные занятия с коррекционными педагогами, логопедами, психологами.
- Квалифицированный уход. Ввиду умственной отсталости пациенты не могут проживать самостоятельно. Уход за ними осуществляют родители с участием медицинского персонала, также рассматривается вариант пребывания больных в специализированных интернатах.
- Диспансерное наблюдение. Пациенты с синдромом Смита-Лемли-Опица должны регулярно проходить медицинские осмотры у ортопеда-травматолога, невролога, уролога и других специалистов. Частота обследований определяется тяжестью клинических симптомов.
Хирургическое лечение
Для устранения аномалий лицевого черепа, синдактилии, гипоспадии проводятся пластические и челюстно-лицевые вмешательства. Объем и сроки операции подбираются с учетом степени тяжести фенотипических изменений. Коррекция врожденных пороков сердца выполняется по стандартным протоколам в зависимости от характера аномалии, наличия расстройств кровообращения. Сложные пороки требуют вмешательства кардиохирургов уже на первом году жизни ребенка.
Прогноз и профилактика
При своевременной диагностике синдрома и раннем начале комплексной терапии прогноз относительно благоприятный. Врачам удается ускорить психомоторное развитие, устранить аномалии строения скелета, скорректировать соматические патологии. Однако низкий рост, умственная отсталость, проблемы с фертильностью остаются на всю жизнь. Профилактика предполагает генетическое консультирование носителей мутировавшего гена перед планированием беременности.
1. Федеральные клинические рекомендации по диагностике и лечению синдрома Смита-Лемли-Опица/ Л.П. Назаренко. — 2015.
2. Врожденные пороки развития. Синдром Смита-Лемли-Опица/ О.А. Милованова, Н.В. Чернышева, А.И. Чубарова, Л.В. Калинина// Неврологический журнал. — 2013. — №3.
3. Случай синдрома Смита-Лемли-Опица/ Л.И. Ширяева, А.М. Поздняков, Е.А. Соляник// Научно-медицинский вестник Центрального Черноземья. — 2006. — №24.
4. Синдром Смита-Лемли-Опица у детей/ А.Н. Семячкина, П.В. Новиков, М.И. Яблонская, М.Б. Кубатов// Российский вестник перинатологии и педиатрии. — 2006. — №3.
Синдром Смит-Магенис
Синдром Смит-Магенис — это редкое генетическое заболевание, которое возникает при микроповреждении короткого плеча 17-й хромосомы. Патология проявляется множественным врожденными пороками развития, среди которых преобладают аномалии лицевого скелета, а также интеллектуальными нарушениями, разнообразными поведенческими особенностями. Для диагностики синдрома Смит-Магенис назначается молекулярно-генетическое тестирование, неврологический обследование, методы инструментальной визуализации. Больным требуется помощь логопедов, реабилитологов, коррекционных педагогов, по показаниям проводится лечение соматических осложнений.
Синдром назван в честь американских врачей ‒ клинициста Энн Смит и цитогенетика Эллен Магенис, которые в 1980 г. впервые описали характерный симптомокомплекс у группы детей. Ранее считалось, что заболевание встречается с частотой 1:25000, но с развитием молекулярно-генетических методов диагностики подтвердилась более высокая распространенность — 1:15000. Чаще всего патология наблюдается у детей с задержкой психического развития — 1 случай на 569 пациентов.
Заболевание возникает вследствие спонтанной хромосомной аномалии — делеции 17 хромосомы в локусе 17p11.2, которая предполагает потерю до 4 млн. нуклеотидных пар. До 25% клинических случаев синдрома Смит-Магенис составляют атипичные микроделеции, которые выражаются в утрате меньшего участка короткого плеча хромосомы. Болезнь носит спорадический характер, пока ученым не удалось установить типичные закономерности и провоцирующие факторы.
Развитие характерной для синдрома симптоматики прежде всего связывают с нарушением гена RAI1, который является индуктором ретиноловой кислоты. Ученые обсуждают его участие в дифференцировке тканей, что объясняет формирование множественных врожденных пороков. Также RAI1 предположительно используется при дифференцировке нейронов. Некоторые психические проявления синдрома Смит-Магенис вызваны дефектом синтеза и деградации меланина.
Дети имеют типичные особенности внешности, которые обнаруживаются уже в младенческом возрасте. Обычно у них широкая квадратная форма головы, выпуклый лоб, глубоко посаженные глаза с монголоидным разрезом. Также для синдрома характерно недоразвитие средней зоны лица, короткий нос с вывернутыми ноздрями, шатровидная верхняя губа при относительно маленькой нижней челюсти. У детей непропорционально короткие руки и ноги, с раннего возраста отмечается задержка роста.
Специфическим диагностическим признаком синдрома Смит-Магенис являются нарушения сна. На первом году жизни наблюдается патологическая сонливость, такие дети много спят днем, их приходится будить для очередного кормления. Со временем нарушаются циркадные ритмы, преобладает поздний отход ко сну с частыми ночными пробуждениям. С возрастом пациент все больше спит днем, не может заснуть ночью.
Психоневрологические изменения включают умственную отсталость умеренной степени тяжести, возникающую практически у всех больных. Характерна задержка психоречевого развития, затруднения при произношении слов, дефицит внимания с гиперактивностью. До 30% пациентов имеют неврологические нарушения: расстройства координации движений, периферическую нейропатию, судорожные приступы.
Болезнь Смит-Магенис сопровождается специфическим поведенческим профилем. Дети страдают стереотипным поведением, проявляют дезадаптивные черты личности — вспышки гнева, непослушание, постоянное требование внимания к себе со стороны взрослых. Некоторые больные склонны к самоповреждению: кусают, щиплют или бьют себя, вводят инородные предметы в физиологические отверстия тела.
Большинство детей имеют мышечную гипотонию с рождения, вследствие чего возникают трудности вскармливания, нарушения глотания, отказ от пищи. В 80% случаев синдром Смит-Магенис ассоциирован с офтальмологическими проблемами: дисплазией радужки, микрокорнеа, пятнами Брушфильда. Более 60% больных страдают хроническими отитами, кондуктивной или нейросенсорной тугоухостью. У половины пациентов диагностируется гиперхолестеринемия.
Пороки сердца встречаются в 25% случаев синдрома. Наиболее распространены дефекты межпредсердной и межжелудочковой перегородок, стеноз трикуспидального клапана, тетрада Фалло. До 15% детей страдают от аномалий мочевыводящих путей, которые включают унилатеральную агенезию почек, удвоение мочеточников, увеличение линейных размеров органов.
Обследование осуществляет мультидисциплинарная команда специалистов с участием педиатра, детского невролога, психиатра, генетика. Синдром Смит-Магенис имеет патогномоничные фенотипические и поведенческие признаки, при выявлении которых устанавливается предварительный диагноз. Для верификации патологии используются следующие методы:
- Генетический анализ. Для обнаружения мутации применяется FISH гибридизация со специфическими зондами, которая определяет патологию с точностью 96-100%. Если тест не показал синдром Смит-Магенис при наличии типичных клинических признаков, выполняются дополнительные молекулярные анализы.
- Пренатальная диагностика. При отягощенном семейном анамнезе или настораживающих результатах УЗИ для исключения синдрома возможно исследование путем амниоцентеза во втором триместре беременности. Учитывая спонтанный характер мутации, такая диагностика производится редко.
- Неврологический осмотр. Пациенту требуется полное обследование с оценкой рефлексов, мышечного тонуса, проб на координацию движений. Обязательно проводятся тесты на уровень интеллекта, степень развития когнитивных функций. Зачастую осмотр дополняется консультацией психиатра.
- Инструментальная визуализация. Методы исследования назначаются дифференцировано. Может потребоваться офтальмоскопия, аудиометрия, исследование вызванных слуховых потенциалов. Для выявления соматических нарушений, типичных для клинической картины синдрома, рекомендованы эхокардиография, УЗИ брюшной полости и забрюшинного пространства.
Лечение синдрома Смит-Магенис
Показано ежегодное комплексное обследование состояния здоровья для контроля динамики нарушений. Больному подбирается индивидуальная программа симптоматической терапии. Медикаментозные средства назначаются редко, в основном для коррекции бессонницы, а психотропные препараты не используются из-за риска усугубления психических проблем. Лечение включает следующие направления:
- Логопедическая помощь. В раннем возрасте занятия с логопедом необходимы для улучшения состояния артикуляционного аппарата и структур полости рта, что облегчает кормление, создает условия для развития речи. В дальнейшем требуется продолжительная коррекция для усовершенствования вербальных навыков.
- Эрготерапия. Частые проблемы с мышечным тонусом и координацией движений требуют участия в программе лечения реабилитологов, которые помогают восстановить моторные функции, развить мелкую моторику, усовершенствовать зрительную и слуховую перцепцию.
- Коррекционные программы. Дети с болезнью Смит-Магенис хорошо учатся в небольших тихих коллективах до 5-7 человек, где каждый получает достаточное внимание и поддержку от преподавателя. Учебные программы подбираются с учетом степени интеллектуальных нарушений.
При осложненном течении синдрома схема лечения расширяется. Неврологические расстройства требуют применения физиотерапии, упражнений ЛФК, противоэпилептических препаратов. Для нормализации зрения некоторым необходима оптическая коррекция, исправление косоглазия. Хирургическое лечение пороков сердца осуществляется в соответствии с выявленной аномалией.
При своевременном начале комплексной терапии синдрома Смит-Магенис удается значительно развить моторные и речевые навыки пациента, адаптировать его к жизни в социуме. Прогноз относительно благоприятный при пожизненной реабилитации, социально-психологической поддержке. Специфические меры профилактики синдрома не разработаны, что обусловлено его спорадическим характером.
1. Smith-Magenis Syndrome / Smith, A. C., & Gropman, A. L. // Cassidy and Allanson's Management of Genetic Syndromes. — 2021.
2. Синдром Смит-Магенис: клиникогенетическая характеристика и молекулярно-цитогенетическая диагностика/ О.М. Хурс, Н.В. Румянцева, Л.В. Исакович, О.Л. Зобикова// Педиатрия. Восточная Европа. — 2015. — №1.
Синдром Паллистера-Киллиана ( Синдром мозаичной изохромосомы 12р , Тетрасомия 12р )
Синдром Паллистера-Киллиана — это крайне редкое генетическое заболевание, которое возникает при тетрасомии по 12-й хромосоме. Его развитие связывают с Х-рецессивным типом наследования. Состояние проявляется множественными патологиями перинатального периода, тяжелыми формами умственной отсталости, разнообразными врожденными аномалиями внутренних органов и стигмами дизэмбриогенеза. Диагностика синдрома Паллистера включает генетическое тестирование, инструментальную визуализацию (УЗИ, КТ, МРТ), неврологическое обследование. Пациентам назначается поддерживающее лечение, комплексная программа реабилитации, регулярное диспансерное наблюдение.
Синдром Паллистера (Паллистера-Киллиана) имеет синонимичные названия тетрасомия 12р, синдром мозаичной изохромосомы 12р. Он встречается крайне редко, в медицинской литературе есть данные о 30 случаях болезни, причем 6 пациентов проживают в Российской Федерации. Патология впервые описана в 1977 г. ученым Ф. Паллистером. В 1981 г. М. Теслер-Никола и В. Киллиан независимо друг от друга более подробно охарактеризовали клиническую картину синдрома. Первый пренатально диагностированный случай заболевания датируется 1985 г.
Болезнь возникает вследствие редкого мозаичного хромосомного расстройства, при котором в генетическом материале клетки образуются 4 копии короткого плеча 12 хромосомы вместо двух, которые должны присутствовать в норме. Эти дополнительные копии, как правило, расположены в одной из хромосом (изохромосома). Предполагается, что у синдрома Паллистера Х-сцепленный рецессивный тип наследования, но данных для подтверждения этой гипотезы недостаточно.
Заболевание появляется в результате генных нарушений, которые вызваны присутствием мозаичного клона mos 47,+ i(12)(p10)/46. Мутация формируется при нерасхождении сестринских хроматид в фазе мейоза II при образовании женских половых клеток. После оплодотворения такой яйцеклетки наблюдается постзиготическое ошибочное деление центромеры и частичная ее потеря при последующих митозах, поэтому только часть клеток тела ребенка имеют тетрасомию 12р.
Синдром Паллистера — тяжелая болезнь, которая может завершаться внутриутробной гибелью плода. В остальных случаях в анамнезе определяются патологии течения беременности в виде раннего токсикоза, угрозы прерывания на поздних сроках, задержки внутриутробного развития плода. У новорожденных отмечаются неврологические осложнения вследствие гипоксически-ишемической энцефалопатии, проблемы с самостоятельным дыханием, средняя или низкая оценка по шкале Апгар.
Больные с синдромом Паллистера имеют специфические особенности лицевого скелета: выступающий лоб с завышенной линией роста волос, низко посаженные деформированные уши, широкое расстояние между глазами. Также встречаются маленький вздернутый нос, вывернутые ноздри, большое расстояние от нижнего края ноздрей до верхней губы. На коже тела выявляются очаги депигментации.
К характерным особенностям синдрома относят диспропорциональное развитие конечностей с укорочением их проксимальных отделов, что сопровождается контрактурами суставов, стойким ограничением объема активных движений. Часто беспокоят проблемы со слухом и зрением. Для синдрома Паллистера-Киллиана типичны пороки сердца (дефект межжелудочковой перегородки, коарктация аорты, открытый Боталлов проток), диафрагмальная грыжа.
При тетрасомии 12р отмечается задержка психомоторного развития, умственная отсталость. Дети поздно начинают переворачиваться, сидеть и ползать, зачастую они не говорят даже в возрасте 1-3 лет, а только произносят отдельные звуки (гуление) или слоги. Наблюдается отставание в росте и физическом развитии, пациенты плохо набирают массу тела, имеют астеническое телосложение. Неврологические нарушения проявляются эпилепсией, мышечной гипотонией, тремором.
У страдающих болезнью Паллистера выявляются множественные пороки на фоне тяжелого неврологического дефицита, что обуславливает раннюю инвалидизацию таких пациентов. Осложнения синдрома связаны с задержкой речи и психического развития, из-за чего больным нужен постоянный уход, квалифицированная медицинская помощь. Неблагоприятный исход обычно наблюдается при тяжелых врожденных пороках, которые с трудом поддаются хирургической коррекции.
Своевременная постановка диагноза затруднена, что обусловлено большой редкостью синдрома Паллистера, наличием тканеспецифичного мозаицизма, что создает сложности при генетических анализах. Перед посещением генетика такие дети зачастую проходят осмотр у большого круга специалистов: педиатров, неврологов, кардиологов, эндокринологов. Для диагностики синдрома Паллистера назначаются следующие методы:
- FISH-анализ. Такой метод генетического тестирования является наиболее точным для определения тетрасомии 12р. Для исследования используются клетки буккального эпителия или культура фибробластов, среди которых до 50-65% имеют характерную мутацию. При анализе лимфоцитов только у 2% клеток обнаруживается дополнительное короткое плечо 12-й хромосомы.
- Консультация невролога. Больным рекомендован детальный неврологический осмотр с оценкой рефлекторной возбудимости, мышечного тонуса, уровня развития психомоторных функций. Обязательно проверяются органы чувств, поскольку у таких детей часто возникают тугоухость, снижение зрения.
- Инструментальные методы. Для выявления сопутствующих врожденных пороков выполняется комплексное обследование с применением УЗИ органов брюшной полости, рентгенографии грудной клетки, эхокардиографии. По показаниям проводится нейросонография, КТ или МРТ головного мозга.
Лечение синдрома Паллистера
Специальные методы лечения синдрома Паллистера пока не разработаны. Основу терапии составляют поддерживающие мероприятия, которые уменьшают клинические проявления болезни, стимулируют формирование отсутствующих психомоторных навыков, помогают адаптировать больного к жизни с инвалидностью. План терапии синдрома включает несколько направлений:
- Метаболические препараты. Лекарства, стимулирующие энергетический обмен, а также витаминно-минеральные комплексы и гомеопатические средства назначаются для поддержки работы миокарда, улучшения функционирования нервной системы.
- Реабилитация. Пациентам показана комплексная реабилитационная программа с участием инструкторов ЛФК, специалистов по механотерапии, дефектологов. Такие мероприятия направлены на развитие у детей устной речи, коммуникативных навыков, улучшения моторных функций.
Оперативное лечение
Сердечные пороки, встречающиеся при синдроме, требуют оперативной коррекции, которая в основном проводится в первый год жизни ребенка. Сроки оказания кардиохирургической помощи определяются тяжестью нарушений кровообращения.
С учетом отсутствия этиопатогенетических методов терапии синдрома Паллистера, тяжести клинической симптоматики и формирования инвалидности в раннем возрасте, прогноз неблагоприятный. Поскольку цель проводимых лечебных мероприятий — улучшение качества жизни пациентов, при их успешной реализации удается значительно развить психомоторные навыки. Специфическая профилактика синдрома отсутствует.
1. Реабилитация больных с синдромом Паллистера-Киллиана с учетом особенностей их метаболизма — клинические наблюдения/ Ю.Б. Гречанина, И.А. Максютина, М.Б. Грузкова// Клиническая генетика и перинатальная диагностика. — 2018. — №1(4).
2. Клинический случай синдрома Паллистера-Киллиана у ребенка/ Т.В. Судовская, Н.Ш. Кокоева, Ю.А. Бобровская, А.А. Макарова// Российская педиатрическая офтальмология. — 2017. — №3.
3. Синдром Паллистера-Киллиана: особенности пре- и перинатальной диагностики/ Н.В. Шилова, Ю.О. Козлова, Н.А. Демина, М.С. Петухова// Медицинская генетика. — 2012. — №11(3).
4. Синдром Паллистера-Киллиана: сонографическая, молекулярно-цитогенетическая и патоморфологическая диагностика/ А.Д. Политыко, И.В. Новикова, Э.И. Мараховская, В.Н. Кошкина// Медицинская генетика. — 2009. — №8.
а) Терминология:
1. Сокращения:
• Синдром Смита-Лемли-Опица (ССЛО)
2. Синонимы:
• Синдром RSH (начальные буквы фамилий первых трех пациентов, описанных Опицем и соавт.)
• CCITO/RSH-синдром
3. Определения:
• Нарушение биосинтеза холестерина, характеризующееся ЗРП, множественными врожденными аномалиями развития и отставанием в умственном развитии:
о ССЛО I и II в ранних литературных источниках: представители фенотипического спектра одного и того же заболевания
б) Лучевая диагностика:
1. Общие сведения:
• Критерии диагностики:
о Тяжелая форма ЗРП на ранних сроках беременности в сочетании с пороками сердца, полидактилией, наличием гениталий промежуточного типа, определяемыми при УЗИ во II триместре

(Слева) УЗИ плода с тяжелой формой ССЛО в III триместре, сагиттальная плоскость. Определяются выраженный отек кожи, короткий вздернутый нос и микрогнатия. Также присутствует тяжелая форма микроцефалии.
(Справа) Клинические признаки ССЛО. Лицевая дизморфия (расщелина нёба и губы) и широкая переносица. Сложная синдактилия и инсерционная полидактилия.
2. УЗИ при синдроме Смита-Лемли-Опица у плода:
• Характерно увеличение ТВП при УЗИ в I триместре
• Аномалии ЦНС:
о Микроцефалия
о ГПЭ
о Вентрикуломегалия
о Гипоплазия мозжечка
о АМТ
• Пороки сердца:
о Открытый АВ-канал
• Аномалии развития мочеполовой системы:
о Гениталии промежуточного типа
о Кистозная дисплазия почек
• Постаксиальная полидактилия; Y-образная синдактилия пальцев стоп
• Краниофациальные аномалии:
о Гипертелоризм
о Короткий вздернутый нос, ноздри, вывернутые кпереди
о Расщелина нёба
о Микрогнатия, уменьшенное ротовое отверстие
• Водянка плода
3. Рекомендации по лучевой диагностике:
• Предпочтительный метод исследования:
о 3D УЗИ позволяет уточнить пороки развития лица и конечностей
о Возможна диагностика в I триместре, особенно в семьях, относящихся к группе высокого риска
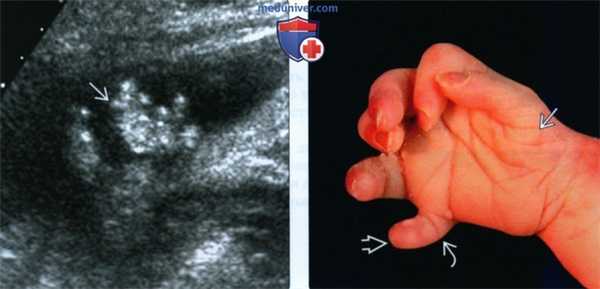
(Слева) При УЗИ плода с ССЛО в III триместре определяется постаксиальная полидактилия. В ходе исследования выяснилось, что пальцы неподвижны.
(Справа) Кисть этого же новорожденного: гексадактилия и постаксиальная полидактилия. Обращает внимание положение пальца во время пренатального УЗИ. Отмечается гипоплазия мышц тенара, а также единственная сгибательная борозда шестого пальца.
в) Дифференциальная диагностика синдрома Смита-Лемли-Опица у плода:
1. Анеуплоидия:
• Т13:
о ГПЭ
о Пороки сердца
о Омфалоцеле
о Расщелина верхней губы и нёба
о Постаксиальная полидактилия
о Крипторхизм
• Т18:
о ЗРП
о Пороки сердца
о Скрещенные пальцы кисти, «стопа-качалка»
о Дефекты радиального луча
о Расщелина верхней губы и нёба
• Триплоидия:
о ЗРП
о Синдактилия II-III пальцев стопы, III—IV пальцев кисти или стопы
о Аномалии развития мочеполовой системы
о Различные аномалии ЦНС
• Делеция 10q:
о Выраженное недоразвитие половых желез
2. Гидролетальный синдром:
• Гидроцефалия
• Пороки сердца
• Расщелина верхней губы и нёба
• Полидактилия
• Крипторхизм
• Укорочение конечностей
3. Псевдотрисомия 13:
• ГПЭ
• Постаксиальная полидактилия
• Гениталии промежуточного типа
• Нормальный кариотип
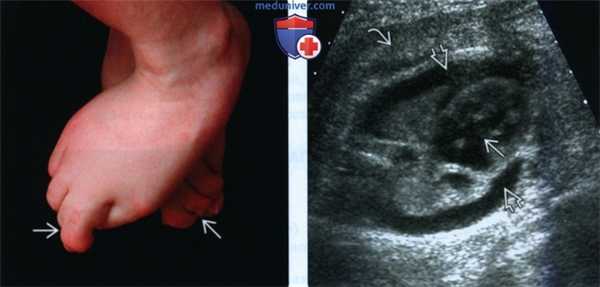
(Слева) Типичная Y-образная синдактилия II-III пальцев у молодого человека с ССЛО. Положение стопы свидетельствует об отсутствии ответа на тактильную стимуляцию, что характерно для ССЛО.
(Справа) УЗИ грудной клетки плода с тяжелой формой ССЛО, поперечная плоскость. Визуализируются двусторонний плевральный выпот и значительное утолщение кожи. Отмечается артериовенозный дефект перегородки. Аутопсия показала однодольное строение легких.
г) Патологоанатомические особенности:
1. Общие сведения:
• Этиология:
о Нарушение биосинтеза холестерина:
- Мутации гена 3-β-гидроксистерол-5-7-редуктазы (DHCR7), катализирующей окончательный этап биосинтеза холестерина, редукцию 7-дегидрохолестерина (7-ДГХ) до холестерина
- Клиническая диагностика ССЛО подтверждается биохимическим исследованием - находят повышение уровня 7-ДГХ в сыворотке крови и тканях:
Уровень холестерина, как правило, снижен, однако у 10% больных может быть в пределах нормы
- Стеролы играют главную роль в образовании миелина, белков ЦНС, мембран:
Изменение профиля стеролов приводит к нарушениям умственного развития и двигательной активности
- Функционирование сигнальных путей Sonic hedgehog и PATCH (сигнальные белки эмбриона) зависит от синтеза холестерина:
Нарушения, связанные с ГПЭ
- Снижение выработки тестостерона и эстрогенов приводит к недоразвитию половых желез у плодов мужского пола:
Во время беременности отмечается низкий уровень свободного эстриола сыворотки крови матери (MSuE3)
о Носительство можно установить с помощью исследования на мутации:
- Установить носительство по анализу уровня холестерина и 7-ДГХ невозможно из-за широкого диапазона нормальных значений
• Генетические факторы:
о Аутосомно-рецессивный тип наследования
о Секвенирование DHCR7 выявляет 96% известных мутаций
2. Гистологические особенности:
• Гигантские клетки панкреатических островков
• Гипоплазия тимуса
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Краниофациальные аномалии: микроцефалия (90%), узкое бифронтальное расстояние, птоз (60%), антимонголоидный разрез глаз, вывернутые кпереди ноздри, расщелина нёба (37-52%), кисты языка, низкорасположенные ушные раковины
о Аномалии мочеполовой системы (90%): инверсия пола у плодов мужского пола или гениталии промежуточного типа, микропения, гипоспадия, агенезия почек, кистозная дисплазия почек, гидронефроз
о Рост: задержка роста плода и новорожденного
о Пороки развития конечностей: постаксиальная полидактилия (50%), Y-синдактилия II-III пальцев стоп (95%), высокая частота извитых папиллярных линий
о Пороки сердца (38%): дефект АВ-канала, аномальный легочный венозный возврат
• Другие субъективные и объективные симптомы:
о Средняя или тяжелая форма нарушения умственного развития
о Характерный поведенческий фенотип: аутизм, членовредительство, отвращение к пище, чрезмерная тактильная чувствительность, нарушения сна, сутулость, раздражительность, нередко предельная
о Дисфункция надпочечников, болезнь Гиршпрунга, аноректальная атрезия
3. Естественное течение и прогноз:
• Тяжелая форма заболевания, проявившаяся в перинатальном периоде, как правило, летальна
• У выживших отмечаются нарушение интеллектуального развития от среднего до тяжелого, многочисленные хронические заболевания, тяжелые поведенческие расстройства
• Для умеренно тяжелого фенотипа характерны поздняя диагностика и более легкое течение
• Пренатальный уровень 7-ДГХ прямо пропорционален тяжести клинического течения
• Тяжесть клинических симптомов в постнатальном периоде обратно пропорциональна уровню холестерина в плазме крови или отношению уровня холестерина к общему уровню стеролов
4. Лечение синдрома Смита-Лемли-Опица у плода:
• Возможна пренатальная диагностика:
о Анализ уровня стерола в околоплодных водах во II триместре (отношение 7-ДГХ/общий уровень стеролов)
о Концентрация 7-ДГХ в ткани ворсин хориона
о Молекулярно-генетический анализ биоматериала, полученного с помощью биопсии ворсин хориона или амниоцентеза
о Преимплантационная генетическая диагностика возможна, если известна конкретная мутация
о Определение уровня стеролов в моче матери (экспериментальный метод)
• Генетическое консультирование с обсуждением пренатального диагноза
• Предлагают прерывание беременности
• Описаны случаи пренатальной терапии:
о Внутрисосудистые и интраперитонеальные инфузии свежезамороженной плазмы
о Нормализуется уровень холестерина в плазме крови
о Долгосрочный прогноз не изменяется, но эти случаи демонстрируют возможность внутриутробного лечения
о В другом случае прием матерью пищевых добавок с холестерином оказался менее эффективным:
- Полученный с пищей холестерин не проникает через гематоэнцефалический барьер и, вероятнее всего, не влияет на развитие ЦНС плода
• Прием пищевых добавок с холестерином и желчными кислотами новорожденным:
о Результаты варьируют
о В некоторых выборках описано улучшение поведения, приема пищи и роста
о Наилучший прогностический фактор потенциала развития - исходный уровень холестерина до лечения
о Проспективных рандомизированных контролируемых исследований к настоящему времени не проводилось в силу редкой встречаемости синдрома:
- Свидетельства о пользе лечения основаны на данных, полученных на малых выборках, и описаниях клинических случаев
• Стресс-дозы стероидов при хирургической патологии, тяжелом состоянии
• Необходимо избегать:
о Введения галоперидола
о Воздействия солнца
е) Особенности диагностики. Признаки, учитываемые при интерпретации изображений:
• Низкий или неопределяемый уровень свободного эстриола (MSuE3) в сыворотке крови матери является показанием к тщательному УЗИ в отношении характерных аномалий развития плода:
о Диагноз можно подтвердить с помощью амниоцентеза или биопсии ворсин хориона
ж) Список использованной литературы:
1. Kanungo S et al: Sterol metabolism disorders and neurodevelopment - an update. Dev Disabil Res Rev. 17(3): 197-210, 2013
2. Lee RW et al: Brain magnetic resonance imaging findings in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 161 A( 10):2407—19, 2013
3. Diaz-Stransky A et al: Cognitive and behavioral aspects of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 160C(4):295-300, 2012
4. Nowaczyk MJ et al: Smith-Lemli-Opitz syndrome: phenotype, natural history, and epidemiology. Am J Med Genet C Semin Med Genet. 160C(4):250-62, 2012
5. Opitz JM et al: The RSH/«Smith-Lemli-Opitz» syndrome: historical footnote. Am J Med Genet C Semin Med Genet. 160C(4):242-9, 2012
6. Orth M et al: Cholesterol: its regulation and role in central nervous system disorders. Cholesterol. 2012:292598, 2012
7. Svoboda MD et al: Treatment of Smith-Lemli-Opitz syndrome and other sterol disorders. Am J Med Genet C Semin Med Genet. 160C(4):285-94, 2012
8. Waterham HR et al: Mutational spectrum of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 160C(4):263-84, 2012
9. Cohen MM Jr: Hedgehog signaling update. Am J Med Genet A. 152A(8):1875-914, 2010
10. Porter FD et al: Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 52(l):6-34, 2010
11. Tint GS et al: Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 330(2): 107-13, 1994
- Вернуться в оглавление раздела "Акушерство."
Редактор: Искандер Милевски. Дата обновления публикации: 20.11.2021
Читайте также:
- Методы обследования гонадной стромальной опухоли яичка
- Диагностика коклюша. Принципы микробиологической диагностики коклюша. Выделение возбудителя коклюша. Идентификация возбудителя коклюша.
- Регуляция секреции поджелудочной железы. Этапы панкреатической секреции
- Происхождение первично-множественных опухолей. Морфология первично-множественного рака
- Осложнения болезни Вильсона-Коновалова. Клиника гемолитического осложнения Вильсона-Коновалова