Синдром Сотоса - этиология, клиника, диагностика
Добавил пользователь Alex Обновлено: 08.01.2026
В практической деятельности педиатр нередко наблюдает больных со значительными отклонениями в физическом развитии. В большинстве обращений эти изменения касаются низкорослости и довольно часто обусловлены наследственными факторами. Но особого внимания зас
Weaver syndrome in a 16-year-old child (clinical case description) / T. A. Bokova, D. A. Kartashova, Yu. Yu. Kotalevskaya
In their practice, pediatricians often observes patients with significant disorders in their physical development. In most cases, these changes are related to dwarfism and quite often conditioned by hereditary factors. But special attention should be paid to children with high anthropometric parameters. The article presents a case of own observation of extremely rare combination of tallness and congenital disorders in development – Weaver syndrome in a 16-year-old adolescent.
В 1974 г. американские генетики, возглавляемые известным ученым, профессором медицинской и молекулярной генетики Медицинской школы Университета Индианы D. Weaver, впервые опубликовали случай данной патологии у двух мальчиков [1]. До настоящего времени в мире описано не более 50 наблюдений этого синдрома. Из ныне живущих больных с синдромом Уивера известна 22-летняя турчанка Румейса Гелги (Rumeysa Gelgi), которая занесена в книгу рекордов Гиннеса как самая высокая женщина в мире. Ее рост составляет 2 метра 15 см. В РНПЦ «Мать и дитя» Минздрава Республики Беларусь в 2013 г. опубликовано 5 случаев наблюдения данного синдрома [2]. В отечественной литературе найдено описание клинического примера 5-летней девочки, наблюдавшейся в Московском НИИ педиатрии и детской хирургии Минздрава РФ в 2000 г. [3].
Тип наследования синдрома Уивера до сих пор остается неясным. Большинство описанных случаев были спорадическими. Проявления синдрома Уивера характеризуются вариабельной экспрессивностью и большей частотой среди лиц мужского пола.
Диагностика заболевания состоит из следующего симптомокомплекса: высокие показатели физического развития (SDS роста > 2), типичные краниофасциальные симптомы, низкий хриплый голос, изменения опорно-двигательного аппарата, поражение органов зрения, возможна задержка психомоторного и речевого развития, часто нормальный уровень соматомедина С и гормона роста, костный возраст соответствует паспортному или опережает его.
Генетическая основа синдрома Уивера длительное время оставалась неизвестной. В 2003 г. J. Douglas с соавт. описали мутации в гене NSD1, отвечающем за развитие синдрома Сотоса, у 3 пациентов из 7 с диагнозом «синдром Уивера» [4]. Однако в 2012 г. было проведено полное секвенирование экзома неродственным пациентам с синдромом Уивера и в результате выявлены мутации в гене EZH2, расположенном на 7-й хромосоме [5].
Дифференциальная диагностика проводится с другими заболеваниями, сопровождающимися макросомией и аномалиями развития: в первую очередь с синдромом Сотоса, синдромом Видемана–Беквита, синдромом МОМО, синдромом Симпсона–Голаби–Бемеля, полисомией Y, синдромом Вермера, синдромом Карнея, синдромом Клайнфельтера, синдромом Марфана, гомоцистинурией, синдромом Пайла, синдромом Маршалла–Смита и другими.
Клиническое наблюдение
Мальчик поступил в педиатрическую клинику МОНИКИ впервые в возрасте 15 лет с жалобами на высокий рост (за последний год прирост 15 см), увеличение размера ноги за год с 44 до 48, деформацию позвоночника и грудной клетки, ухудшение зрения.
Ребенок от второй беременности (первый ребенок здоров), протекавшей на фоне повторных угроз прерывания на ранних сроках. Роды срочные, самопроизвольные. Масса при рождении 4200 г, длина 57 см. С рождения физическое развитие гармоничное, высокое. Психомоторное развитие и половое развитие соответствуют возрасту. Наблюдается офтальмологом по поводу гиперметропии высокой степени обоих глаз, хирургом-ортопедом по поводу нарушения осанки, деформации грудной клетки, плоскостопия, челюстно-лицевая хирургия по поводу микрогнатии (получает лечение в виде дистракционного остеосинтеза нижней челюсти с двух сторон) и ортодонтом по поводу нарушения прикуса и роста зубов. Рост отца и матери ребенка 172 см. У родственников мужского пола по линии матери рост 180 см — 2 м.
С рождения отмечались большие прибавки роста. При обследовании в 2 года — физическое развитие на 4 года. Общеклинические анализы в норме. Гормон роста в пределах референсных значений. Костный возраст на 3–4 года. МРТ головного мозга — без патологии. Ребенок и в детском саду, и в школе был значительно выше своих сверстников.
С 2012 г. появились жалобы на головокружение, головную боль, периодический акроцианоз, потерю сознания при изменении положения тела. При мониторинге артериального давления выявлена склонность к гипотонии. Предъявляемые ребенком жалобы объяснялись отставанием развития сердечной мышцы и сосудистой системы по сравнению со скоростью роста костей, тогда как сама причина такого стремительного роста оставалась неясной.
В 2017 г. мальчик поступил в педиатрическое отделение МОНИКИ с направительным диагнозом: «Синдром высокорослости».
При поступлении рост мальчика рост 196 см, вес 78 кг. Обращали на себя внимание: грубый хриплый голос, лицевые дизморфии (антимонголоидный разрез глаз, удлиненные глазные щели, широкий и короткий нос, широкий длинный фильтр, ретромикрогнатия, большие диспластичные ушные раковины), скелетные деформации (арахнодактилия, конусовидные пальцы кистей, кифоз, сколиоз, деформация грудной клетки) (рис.). По лабораторным данным: общий анализ крови, мочи, кала, развернутый биохимический анализ крови, гормональный профиль (гормоны щитовидной железы, гипофиза, надпочечников, половые гормоны), исследование фосфорно-кальциевого обмена — показатели в пределах референсных значений. ЭКГ, Эхо-КГ, УЗИ брюшной полости, забрюшинного пространства и щитовидной железы — без особенностей. Уровень артериального давления в пределах возрастной нормы. МРТ головного мозга и гипоталамо-гипофизарной области прицельно — без патологии. Рентгенограмма кистей — костный возраст соответствует 15,5–16 годам. Осмотрен совместно с узкими специалистами (эндокринолог, генетик): SDS роста +3,4. Половое развитие соответствует возрасту. Высокорослость. Марфаноподобный синдром.
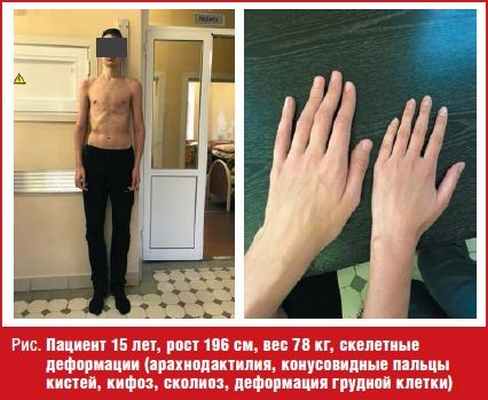
В результате комплексного обследования в клинике эндокринная причина избыточного роста ребенка была исключена, и тогда было принято решение провести углубленное генетическое обследование. Проведен поиск мутаций, ассоциированных с синдромом Марфана и другими наследственными заболеваниями со сходными фенотипическими проявлениями. В результате молекулярно-генетического анализа у мальчика выявлена мутация в 17-м экзоне гена EZH2 в гетерозиготном состоянии. Мутация в гене EZH2 является типичной для синдрома Уивера, однако выявленная у пациента мутация ранее не была описана.
Таким образом, учитывая фенотипические особенности пациента и наличие мутации в гене EZH2, был установлен диагноз «синдром Уивера».
Спустя год ребенок обследован в клинике повторно. Рост 2,01 м (годовая прибавка роста 5 см), вес 86 кг. Размер обуви остался без изменений. В ходе обследования общеклинические анализы, данные гормонального профиля, форфорно-кальциевого обмена, УЗИ брюшной полости, Эхо-КГ, мониторинг артериального давления, МРТ головного мозга, денситометрия в режиме позвоночник и все тело — без патологии. Костный возраст соответствует 16–16,5 годам.
Заключение
Несмотря на то, что все генетически детерминированные синдромы высокорослости встречаются крайне редко, характеризуются схожими проявлениями, не имеют специфического лечения, пациентам необходимо проводить комплексное генетическое обследование. В зависимости от генетического дефекта, различается спектр клинических проявлений различных синдромов, что позволяет прогнозировать в дальнейшем течение заболевания и осуществлять профилактические мероприятия по развитию некоторых осложнений. Так, например, учитывая, что мутации в гене EZH2 являются причиной синдрома Уивера, а ген EZH2 является белком гомеобокс группы, который участвует в поддержании гомеостаза, деления и дифференцировки клеток, соответственно, патологическая экспрессия этого гена у больных синдромом Уивера может быть причиной нейродегенеративных заболеваний и некоторых форм рака, например, нейробластомы. Кроме того, в гене EZH2 описаны мутации в соматических клетках при лейкемии, В-клеточных лимфомах и других онкологических заболеваниях крови, что обуславливает некоторую предрасположенность пациентов с синдромом Уивера к онкологическим заболеваниям [5–7]. Эти сведения могут помочь лечащему врачу в определении тактики наблюдения пациента и осуществления профилактических мероприятий.
Литература
- Weaver D. D., Graham C. B., Thomas I. T., Smith D. W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly // J. Pediatr. 1974; 84: 547–552.
- Ильина Е. Г., Ершова-Павлова А. А. Синдром Вивера в Беларуси // Здравоохранение (Минск), 2013; 3: 62–65.
- Казанцева Л. З., Семячкина А. Н., Новиков П. В., Новикова И. М., Добрынина Э. В. Синдром Вивера у детей // Российский вестник перинатологии и педиатрии. 2000; 2: 55–57.
- Douglas J. et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes // Am J Hum Genet. 2003, Jan; 72 (1): 132–143.
- Gibson, W. T., Hood R. L. et al. Mutations in EZH2 cause Weaver syndrome // Am. J. Hum. Genet. 2012, 90: 110–118.
- Качанов Д. Ю., Шаманская Т. В., Шевцов Д. В. и др. Генетическая предрасположенность к нейробластоме у детей: собственные данные и обзор литературы // Онкопедиатрия. 2016; 3 (4): 277–287.
- Chase A., Cross N. C. Aberrations of EZH2 in cancer // Clin Cancer Res. 2011, May 1; 17 (9): 2613–2618.
Т. А. Бокова 1 , доктор медицинских наук, профессор
Д. А. Карташова
Ю. Ю. Коталевская, кандидат медицинских наук
ГБУЗ МО МОНИКИ им. М. Ф. Владимирского, Москва
Синдром Уивера у ребенка 16 лет (описание клинического случая)/ Т. А. Бокова, Д. А. Карташова, Ю. Ю. Коталевская
Для цитирования: Лечащий врач № 1/2019; Номера страниц в выпуске: 12-13
Теги: синдром Уивера, физическое развитие, отклонения
Синдром Сотоса - этиология, клиника, диагностика
Проблема редких (орфанных) болезней стала настолько актуальной, что привлекает внимание органов государственной власти, медицинской общественности, средств массовой информации, родителей больных детей и родительских ассоциаций не только в нашей стране, но и в мире.
Благодаря росту информированности врачей и развитию ранней диагностики в ближайшие годы количество выявленных пациентов с орфанными заболеваниями в России будет продолжать расти. Соответственно, медицинским учреждениям необходимо наращивать возможности для диагностики, лечения и реабилитации таких пациентов. Этот тезис прозвучал на III Междисциплинарной конференции «Орфанные заболевания. Диагностика. Лечение. Реабилитация», которая прошла в конце ноября в Санкт-Петербурге в ФГБУ «НМИЦ им. В. А. Алмазова» Минздрава России.
Детский лечебно-реабилитационный комплекс (ДЛРК) открылся в 2017 году на базе НМИЦ им. В. А. Алмазова. Кроме наблюдения детей после сложных хирургических операций, специалисты Центра Алмазова занимаются лечением и реабилитацией маленьких пациентов, с рождения страдающих орфанными заболеваниями. На базе ДЛРК сформировался мультидисциплинарный центр по диагностике, лечению и наблюдению пациентов с редкими заболеваниями. Медицинская помощь оказывается не только на стационарном, но и на амбулаторном уровнях.
Сегодня пациентами Центра Алмазова являются дети с такими редкими заболеваниями, как гипофосфатазия (детская форма), мукополисахаридозы, в частности синдром Гурлера и синдром Хантера, синдром апноэ во сне (синдром Ундины), миотония Дюшена-Беккера, пропионовая ацидемия, синдром Сотоса, синдром Криглера-Найяра, спинально-мышечная атрофия и многие другие.
По словам главного врача Детского лечебно-реабилитационного комплекса клиники Института перинатологии и педиатрии ФГБУ «НМИЦ им. В. А. Алмазова» Минздрава России Елены Юрьевны Гуркиной, сегодня количество пациентов с орфанной патологией растет за счет повышения уровня выявляемости таких болезней. «Известных науке орфанных болезней сейчас насчитывается порядка 6000-7000. Но даже если брать те, которыми мы реально занимаемся, а их несколько десятков, то, чем больше мы о них говорим, тем больше о них знают, и это способствует выявлению таких заболеваний -- мы это видим, -- говорит Елена Юрьевна. -- Кроме того, большему выявлению способствует и то, что появляется возможность терапии таких заболеваний, соответственно, есть стимул их искать и находить».
Как утверждают специалисты Центра Алмазова, еще несколько лет назад в клинике учреждения занимались только 7–8 редкими нозологиями. Сегодня же врачи разных специальностей работают с более чем 20 орфанными нозологиями. И их количество постоянно растет. Поэтому в качестве основных тенденций в ближайшее время эксперты прогнозируют расширение компетенций специализированных центров по лечению пациентов с редкими заболеваниями. В первую очередь это означает рост уровня информированности об орфанных заболеваниях всего медицинского сообщества.
Дело в том, что успех лечения орфанного пациента во многом зависит от ранней постановки диагноза: чем раньше обнаружено редкое заболевание, тем больше шансов у пациента своевременно начать необходимое лечение. Это важно еще и потому, что отсутствие необходимого лечения, например, при неверно поставленном диагнозе, ведет к необратимым изменениям в организме, а далее -- к инвалидности и возможной смерти.
Одним из примеров такого заболевания является мукополисахаридоз 2 типа (МПС-2, синдром Хантера), которое является неизлечимой генетической болезнью. До недавнего времени терапии для пациентов с таким диагнозом не существовало, а продолжительность жизни детей с МПС-2 не превышала 10-15 лет. Ситуация в мире изменилась после появления рекомбинантного фермента идурсульфазы (препарат Elaprase). Ферментозаместительная терапия (ФЗТ) с применением этого препарата позволяет затормозить развитие болезни и улучшить состояние больного. Российские пациенты с МПС-2 получили доступ к ФЗТ-препарату идурсульфаза в 2008 году, после его регистрации на территории РФ. А полное и бесперебойное обеспечение препаратом «Элапраза» пациенты с синдромом Хантера получили в 2019 году, после включения мукополисахаридоза 2 типа в федеральное финансирование и включения лекарства в список жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП). Теперь пациенты обеспечиваются лекарством централизованно из федерального бюджета.
Что касается МПС-2, то наблюдение пациентов с синдромом Хантера идет в НМИЦ им. В. А. Алмазова уже более двух лет. Сейчас, по словам главного специалиста -- детского кардиолога Северо-Западного федерального округа Минздрава России, врача-педиатра, врача -- детского кардиолога высшей категории д.м.н. Елены Сергеевны Васичкиной, в Центре Алмазова на лечении находятся 4 пациента с синдромом Хантера: младшему из них 2,9 лет (он уже несколько месяцев получает ФЗТ идурсульфазой), старшему — 16 лет. И, по мнению доктора, можно сказать, что некоторый опыт работы с такими пациентами у специалистов НМИЦ им. В. А. Алмазова уже имеется.
«Пациентов с синдромом Хантера, которые получают ферментозаместительную терапию, необходимо наблюдать продолжительнее время. Но уже сейчас можно отметить, что никаких острых или негативных реакций на препарат у них нет. Когда мы обсуждаем с родителями подходы лечения, мы делам акценты на том, что синдром Хантера -- это прогрессирующее хроническое заболевание, приостановить которое и улучшить прогноз можно только ферментозаместительной терапией», -- добавляет Елена Сергеевна.
Немаловажным фактором для улучшения качества жизни детей с МПС-2 является также компетенция врачей-специалистов, проводящих ферментозаместительную терапию. Здесь важна преемственность, поскольку специалисты из крупных орфанных центров не только сами освоили правильное и безопасное введение инфузий, но и занимаются обучением этому своих региональных коллег.
Подготовку молодых специалистов, как считают в Центре Алмазова, нужно начинать уже на студенческом уровне. Необходимо проводить программы, которые повышают уровень информированности врачей, позволяют своевременно выявлять орфанное заболевание, а затем и обеспечивать пациента необходимой терапией. Специалисты центра надеются, что со временем многие орфанные заболевания станут менее редкими за счет ранней диагностики. А пациенту это сохранит жизнь и позволит оставаться в течение жизни активным членом общества.
Для родителей детей с редкими заболеваниями в Детском лечебно-реабилитационном комплексе Центра Алмазова регулярно проводится Школа «Ребенок с редким заболеванием». Участникам школы рассказывают о течении различных орфанных болезней и их осложнениях, возможностях современной медицины в диагностике и лечении редкой патологии, физической и социальной реабилитации и улучшении качества жизни ребенка и семьи, освещаются аспекты организации медико-социальной помощи и юридической поддержки.
Синдром Сотоса
Синдром Сотоса – редкое генетическое заболевание, которое проявляется разнообразными нарушениями формирования скелета, умственной отсталостью, аномальными чертами лица и диспропорциональностью развития тела. Симптомами синдрома Сотоса являются высокий рост, увеличенный размер черепа, стоп и кистей, макроглоссия, замедленное психомоторное развитие с резким ускорением роста в пубертатный период. Диагностика синдрома Сотоса производится на основании антропометрических данных, результатов рентгенологических исследований, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения данного состояния не существует, применяется симптоматическая терапия и психологические коррекционные мероприятия.
Общие сведения
Синдром Сотоса (синдром церебрального гигантизма) – редкое генетическое заболевание, чаще всего являющееся результатом спонтанных мутаций, приводящее к нарушению развития скелета и центральной нервной системы. Впервые данная патология была описана американским педиатром Хуаном Сотосом в 1964 году, с тех пор заболевание носит его имя. Синдром Сотоса является крайне редким аутосомно-доминантным заболеванием, за всю историю было описано порядка 120 случаев, по этой причине достоверно установить его встречаемость не представляется возможным. Редкость этого состояния стала причиной его довольно медленного изучения – молекулярно-генетические механизмы развития патологии были открыты только в начале XXI века. Удалось не только определить гены, мутации которых приводят к развитию синдрома Сотоса, но и установить тот факт, что обычно такие мутации являются результатом спонтанных генетических дефектов. Данное заболевание чаще поражает мальчиков, однако причины полового распределения на сегодняшний момент неизвестны.
Причины и классификация синдрома Сотоса
По данным современной генетики синдром Сотоса является гетерогенным заболеванием, так как к его развитию могут приводить дефекты различных генов. По этому признаку построена классификация данной патологии, на сегодняшний момент известно два ее типа, которые по своим клиническим проявлениям практически неотличимы друг от друга. Синдром Сотоса первого типа обусловлен мутациями гена NSD1, который располагается на 5-й хромосоме и кодирует последовательность одного из факторов транскрипции. Причиной заболевания могут выступать как точечные мутации гена, так и хромосомные аномалии (делеции) в локусе, где находится NSD1, что приводит к резкому угнетению экспрессии белка или даже отсутствию его выделения. При этом мутация должна находиться в гетерозиготном состоянии (имеет место явление гаплонедостаточности) – по некоторым данным, наличие дефекта обоих аллелей не ведет к синдрому Сотоса, а является летальной мутацией.
Синдром Сотоса второго типа вызывается повреждением гена NFIX, который локализован на 19-й хромосоме. Эта разновидность патологии считается более редким вариантом заболевания. Как и в предыдущем случае, наследуется по аутосомно-доминантному механизму или (намного чаще) возникает в результате спорадических мутаций. Продуктом экспрессии гена NFIX также является транскрипционный фактор (ядерный фактор транскрипции 1), контролирующий активность целого ряда других генов, принимающих участие в развитии центральной нервной системы и других отделов организма. Помимо этого заболевания, повреждения гена NFIX приводят к возникновению такой патологии, как синдром Маршалла-Смита. Как уже было сказано выше, дифференцировать различные типы синдрома Сотоса по клиническому течению не представляется возможным, только молекулярно-генетический анализ способен выявить, какой именно ген был поврежден.
Молекулярные основы патогенеза при различных вариантах синдрома Сотоса также очень сходны. Экспрессируемые генами NSD1 и NFIX транскрипционные факторы регулируют активность целого ряда иных генов, многие из которых принимают активное участие в эмбриогенезе, развитии различных органов и систем. При наличии мутации в гетерозиготном состоянии активность этих факторов становится недостаточной, что нарушает работу контролируемых генов и приводит к возникновению синдрома Сотоса. Выяснено, что форма заболевания первого типа также сопровождается нарушением активности андрогенных рецепторов, а второго типа – эстрогеновых.
Симптомы синдрома Сотоса
Многие проявления синдрома Сотоса можно обнаружить уже при рождении ребенка, что позволяет заподозрить наличие этого генетического заболевания. Как правило, физические показатели новорожденных (длина тела, окружность головы, вес) несколько выше средних значений. Практически всегда при синдроме Сотоса выявляются аномалии развития лица и головы – гипертелоризм, антимонголоидный разрез глаз, макродолихоцефалия, наличие надбровных бугров, прогнатизм. Постоянным признаком заболевания является макроглоссия, из-за которой может значительно затрудняться процесс питания ребенка. Также у новорожденных с синдромом Сотоса может обнаруживаться высокое арковидное нёбо, в крайне редких случаях возникает его расщепление, возможны признаки врожденных пороков сердца.
Рост детей с синдромом Сотоса резко ускорен – особенно интенсивно физические показатели увеличиваются в период 3-4 лет, при этом моторное и психологическое развитие больных отстает от возрастной нормы. Дети с такой патологией позже сверстников начинают приподнимать головку, ходить, достаточно сильно задерживается развитие речи, моторных навыков. Как правило, в период полового созревания рост тела повторно резко ускоряется, однако это ускорение непродолжительно, поэтому больные с синдромом Сотоса в конечном итоге имеют физические показатели на грани верхней границы нормы. Довольно часто при этом заболевании отмечается увеличение относительных размеров кистей и стоп (акромегалия), на ногах иногда определяется синдактилия.
Взрослые больные синдромом Сотоса отличаются высоким ростом, незначительной диспропорциональностью тела, увеличенным размером головы и нижней челюсти. У них также могут диагностироваться искривления позвоночного столба, плоскостопие, искривление ног, однако эти проявления достаточно редки. Умственное развитие при синдроме Сотоса значительно снижено, лица с этим заболеванием начинают поздно говорить, в дальнейшем сохраняется замкнутость, неразговорчивость. Эмоциональная сфера таких больных достаточно нестабильна, в ряде случаев они чрезмерно возбудимы, быстро меняют настроение, иногда могут проявлять признаки агрессии, в том числе и немотивированной.
Диагностика и лечение синдрома Сотоса
Ввиду значительного спектра нарушений и аномалий при синдроме Сотоса для определения этого заболевания используется большое количество разнообразных диагностических техник. К ним относят общий осмотр больных, рентгенографию костей скелета, магнитно-резонансную томографию головного мозга, электроэнцефалографию, молекулярно-генетические анализы. Для оценки нервно-психического развития детей и взрослых с этой патологией используются неврологическое и психологическое исследования. Кроме того, вспомогательную роль в диагностике синдрома Сотоса могут играть анализы для определения уровня гормонов гипофиза в крови, кардиологические исследования, УЗИ внутренних органов. При осмотре выявляются характерные для данного заболевания нарушения – диспропорциональность сложения, превышение возрастных норм физических показателей, изменения лица.
На рентгенограммах определяется опережение костного возраста относительно фактического, может также обнаруживаться искривление позвоночного столба и деформации некоторых других костей. Магнитно-резонансная томография при синдроме Сотоса в некоторых случаях подтверждает недоразвитие мозолистого тела, иногда выраженных анатомических изменений в головном мозге не выявляется. При этом электроэнцефалография всегда свидетельствует о снижении фоновой активности работы нервной системы, есть ЭЭГ-признаки нарушения взаимоотношений между корой и подкорковыми структурами. Молекулярно-генетическая диагностика синдрома Сотоса осуществляется врачом-генетиком и сводится к выявлению мутаций в генах NSD1 или NFIX методом прямого автоматического секвенирования. Иногда для определения этого заболевания выполняют поиск делеций на 5-й хромосоме посредством FISH-технологии.
Для исключения других патологий, сопровождающихся увеличением физических показателей или умственной отсталостью, назначают анализы для определения уровня гормонов гипофиза или надпочечников – при синдроме Сотоса эти показатели остаются в пределах нормы. Электрокардиограмма и ультразвуковые исследования могут выявить аномалии развития сердца и других внутренних органов, что нередко имеет место при данном заболевании. Специфического лечения синдрома Сотоса не существует, применяют симптоматическую терапию. Проводят коррекцию пороков сердца, аномалий внутренних органов. Искривление позвоночного столба и деформации костей могут быть исправлены традиционными ортопедическими методиками. В ряде случаев требуется нейропсихологическая коррекция и (ввиду умственной отсталости) специализированное обучение больных синдромом Сотоса.
Прогноз и профилактика синдрома Сотоса
Как правило, прогноз синдрома Сотоса относительно благоприятный, так как, несмотря на умственную отсталость и изменения скелета, жизни больных ничего не угрожает. При своевременной адекватной психотерапевтической помощи и специализированном обучении лица с этим заболеванием могут себя обслуживать. Ухудшить прогноз синдрома Сотоса может наличие сопутствующих нарушений со стороны внутренних органов – главным образом, врожденных пороков сердца. По некоторым данным, при этой патологии также существует повышенный риск развития некоторых онкологических заболеваний. В крайне редких случаях больные синдромом Сотоса бывают агрессивными, что требует их кратковременной изоляции в специализированных учреждениях и назначения успокоительных препаратов. Профилактика этого заболевания ввиду спонтанности его развития на сегодняшний момент не разработана.
Синдром Сотоса: признаки, лечение, прогноз
В медицинской литературе синдром Сотоса еще известен под названием «церебральный гигантизм». Для него характерны высокий рост в детском возрасте, увеличенный размер головы, различные черепные и лицевые аномалии, а также нарушение речи и интеллекта.
Развитие современной медицины и проведение исследований в области молекулярной биологии и генетики позволили выяснить причины и механизмы развития синдрома, но эффективные методы лечения и профилактики до сих пор не разработаны.
Почему возникает

Синдром Сотоса — врождення патология, причиной которой является генная мутация
Синдром Стотоса относится к числу врожденных генетических заболеваний с аутосомно-доминантным типом наследования, но чаще возникает в результате спонтанных мутаций.
Данная патология может передаваться детям от родителей, но проявляется не у всех особей, что связано с низкой пенетрантностью патологического гена и генетической неоднородностью болезни.
Медицинская наука выделяет 2 типа синдрома, хотя клинически они не различимы.
- Возникновение синдрома 1 типа обусловлено мутацией в гене NSD1 или хромосомной аномалией в локусе, где он находится. Этот ген располагается на 5 хромосоме и кодирует белок, оказывающий влияние на активность андрогенных рецепторов и принимающий участие в регуляции нормального роста и развития.
- Второй тип синдрома связан с мутаций в гене NFIX, локализованном на коротком плече 19 хромосомы. Последний определяет активность белка – активатора фосфопротеинов, участвующего в регуляции транскрипции генов, которые взаимодействуют с эстрогеновыми рецепторами.
И в том, и в другом случае мутации в генах вызывают сбои функционирования многих процессов в организме. При этом изменяется активность целого ряда регуляторных белков и ферментов, что приводит к нарушению роста и развития.
Как проявляется
Признаки заболевания появляются у ребенка еще в период внутриутробного развития. Новорожденные с синдромом Сотоса отличаются большой массой и длиной тела, а также размером головы. В среднем длина тела при рождении составляет 55 см, а масса – более 3900 г. Большая часть детей переносит желтуху в неонатальном периоде. Фенотипические признаки могут варьировать у разных пациентов. Чаще всего у таких детей выявляются:
- непропорционально крупный череп с выпуклой лобной частью и сплюснутыми висками;
- долихоцефалия;
- залысины в лобно-теменных областях;
- антимонголоидный разрез глаз;
- увеличенные челюсти и заостренный подбородок;
- высокое готическое небо;
- грубые черты лица;
- макроглоссия (большой язык);
- гипертелоризм (увеличение расстояния между глазницами); ;
- крупные руки и ноги.
Следует отметить, что это наиболее часто встречающиеся признаки синдрома. Однако у некоторых детей фенотип может быть необычным, например, они могут иметь монголоидный размер глаз или избыточный рост волос на теле.
В дальнейшем у детей отмечаются особенности телосложения:
- низкая масса тела;
- удлиненная шея;
- узкая грудная клетка;
- повышенная разгибаемость суставов;
- большие кисти и стопы;
- иногда – кифосколиоз, плоскостопие.
Нервно-психическое развитие еще в большей мере задерживается, чем моторное. Такие дети поздно начинают говорить. Их речь часто невнятна и состоит из отдельных звуков, слогов, затем реплик. Возможно нарушение фонации, заикание. Характерным проявлением синдрома является умственная отсталость. Однако снижение интеллекта может иметь различную степень выраженности (чаще это легкое или умеренное снижение). Кроме того, развитие синдрома может сопровождаться:
- повышенной раздражительностью;
- агрессивностью;
- замкнутостью; ; ;
- судорогами;
- нарушением координации движений.
Нередко у таких детей нарушается функционирование внутренних органов в результате висцеромегалии и пороков развития (особенно сердечных аномалий).
По мере взросления ребенка с синдромом Сотоса рост его замедляется. У взрослых он не выходит за пределы нормальных значений:
- у мужчин он составляет 184,3 ± 6 см;
- у женщин – 172,9 ± 5,7 см.
Специфические черты лица сглаживаются и становятся менее заметными. Хотя обращает на себя внимание выступающий подбородок. Мышечный тонус нормализуется. Происходит улучшение речевой функции. Половое созревание может наступать относительно рано, хотя остается в пределах нормального диапазона.
У каждого пациента проявления синдрома выражены в разной степени. У некоторых из них возможно достижение нормальных показателей интеллектуального и физического развития во взрослом возрасте. Но большая часть пациентов имеют умственную отсталость и речевые нарушения.
Диагностика
Молекулярно-генетическая диагностика подтвердит диагноз
Диагностика синдрома базируется на характерных клинических признаках. У ребенка четко прослеживается опережение темпов роста и отставание в нервно-психическом развитии, а при осмотре выявляются типичные фенотипические признаки. Для подтверждения диагноза назначается дополнительное обследование, которое включает:
- рентгенографию кистей и определение костного возраста (опережает календарный на 2-4 года);
- определение эндокринного статуса – анализ крови на уровень гормонов (все показатели в пределах нормы);
- молекулярно-генетическая диагностика (выявление мутаций в генах – NSD1, NFIX).
Для оценки функционирования нервной системы и выявления патологических изменений могут применяться следующие методы:
С целью исключения сердечных пороков больным назначается ЭКГ и эхокардиография.
Дифференциальная диагностика проводится с заболеваниями, при которых имеется высокий рост и задержка нервно-психического развития:
- (болеют мужчины, имеющие характерный генотип ХХУ, непропорционально длинные конечности, гипогонадизм и сниженный интеллект);
- гомоцистинурия (нарушение обмена аминокислот; у ребенка выявляется подвывих хрусталика, катаракта, глаукома, деформации конечностей и грудной клетки, частые тромбозы);
- синдром ХУУ (обнаруживается лишняя У-хромосома, аномалии поведения);
- синдром Беквита-Видемана (кроме высокого роста у больных выявляются насечки на мочках ушей, грыжи пупочного канатика, гипогликемические состояния).
Лечение и прогноз
Этиопатогенетическое лечение синдрома не разработано. Пациенты с синдромом Сотоса подлежат наблюдению и реабилитации. При необходимости проводится симптоматическая терапия. Ее целью может быть дегидратация или купирование судорог, улучшение микроциркуляции и трофики в тканях.
Реабилитационные мероприятия заключаются в следующем:
- занятия с логопедом;
- психо-педагогическая коррекция;
- трудовая реабилитация;
- физиотерапия и ЛФК;
- выработка навыков самообслуживания;
- предупреждение формирования стереотипных движений и патологической установки.
Прогноз для жизни при синдроме Сотоса благоприятный. Продолжительность жизни у таких больных не отличается от средних показателей в популяции.
К какому врачу обратиться
Так как основные проблемы пациентов с синдромом Сотоса заключаются в нарушении интеллектуального развития, их наблюдает детский психиатр и невролог. Требуется лечение у логопеда, курсы массажа, занятия у специалиста по лечебной физкультуре, консультация генетика и кардиолога.
Заключение
Проблема диагностики, лечения и реабилитации больных с редкими заболеваниями, в том числе и синдромом Сотоса, является особо актуальной в наше время во всем мире. Их симптомы достаточно специфичны, но с учетом низкой частоты встречаемости не всегда выявляются вовремя. Дети с выраженными признаками синдрома и врожденными пороками развития, как правило, сразу привлекают внимание специалистов. В то время как лица, имеющие более стертую клинику, могут длительное время не получать нужной помощи. Реабилитация таких детей очень важна, ведь она позволяет им адаптироваться к жизни с болезнью, которая, к сожалению, пока неизлечима.
Информативно о синдроме Сотоса (англ. яз.):
Синдром Сотоса
Синдром Сотоса или церебральный гигантизм – это редкая разновидность генетического заболевания, для которого характерно наличие отклонений в формировании скелета, отсталость умственного развития, нарушения лицевых черт и диспропорциональность в развитии отдельных частей тела.
Этот недуг передается по аутосомно-доминантному типу – от родителей детям. Генетикам удалось установить гены, отвечающие за развитие патологии, что упрощает проведение анализа и выявление мутаций.
Наличие отклонений заметны при внутриутробной диагностике или видны визуально после рождения малыша. Обследование пациента может включать в себя общий осмотр, рентгенографию скелета, магнитно-резонансную томографию головы, а также молекулярно-генетический анализ. Комплексное обследование поможет подтвердить недуг и уточнить общую картину нарушений.
Синдром Сотоса не имеет специфической терапии, лечение назначается только симптоматическое, возможна коррекция внутренних аномалий с помощью хирургического вмешательства или использование ортопедических методик. Такие дети требуют специализированного обучения и ухода. Прогноз для жизни – благоприятный.
Этиология
Генетиками была установлена основная причина возникновения заболевания – генные мутации. Такие изменения были выявлены в двух основных генах, изменения в которых привели к катастрофическим последствиям.
Синдром Сотоса у детей проявляется из-за дефекта в следующих генах:
- NSD1 – этот ген относится к первому типу патологии и связан с изменениями в 5-й хромосоме. Мутация такого типа может быть точечной или хромосомной.
- NFIX – это второй тип заболевания, связанный с аномалиями в 19-й хромосоме в гене NFIX. Это более редкая разновидность деформации и чаще появляется из-за спорадических мутаций. В этом случае нарушения могут спровоцировать синдром Маршалла-Смита.
Дифференцировать разновидность мутационных изменений в том или ином гене можно только с помощью проведения лабораторных исследований.
Способ передачи болезни в обеих разновидностях аутосомно-доминантный – от взрослых к детям и характеризуется следующими особенностями:
- в каждом поколении встречаются больные дети;
- болеют в равной степени как женщины, так и мужчины;
- если у больных родителей гомозиготный тип, то вероятность передачи – 100%;
- если родители гетерозиготны, то вероятность передачи – 75%;
- у здоровых родителей вероятность появления больных детей равна 0%.
При планировании беременности стоит пройти процедуру генетического обследования, чтобы выявить или опровергнуть наличие аномалии.
Симптоматика
Синдром Сотоса манифестируется с момента рождения. Чаще всего новорожденные с таким типом патологии значительно крупнее, чем среднестатистический ребенок.
При осмотре младенца отмечается:
- превышение физических показателей;
- аномалии в развитии лица;
- гипертелоризм головы, в этом случае наблюдается ненормальное расстояние между органами зрения;
- антимонголоидный разрез глаз, характерной чертой которого является общинность угла обоих органов зрения;
- макродолихоцефалия – это увеличение размера головы без избыточного скопления церебральной жидкости в желудочках мозга;
- надбровные бугры;
- прогнатизм, в этом случае отмечается противоестественное выступление нижней челюсти вперед.
К основному постоянному признаку недуга относят макроглоссию, из-за чего процесс потребления пищи малышом может сильно затрудняться.
- наличие арковидного неба;
- расщепление неба;
- наличие врожденных пороков в сердце.
У таких детей отмечается ощутимая ускоренность роста, особенно это видно в период четырехлетнего возраста, с отставанием в моторном и психологическом развитии. Ребенок с синдромом Сотоса позже начинает поднимать голову, ходить. У него задерживается развитие моторики и речь.
Повторное ускорение роста отмечается в период полового созревания, однако, это длится недолго и не превышает показатели нормы.
Параллельно может наблюдаться увеличение кистей и стоп.
У взрослых с таким диагнозом визуально присутствует:
- нарушения пропорции тела;
- увеличение головы и нижней челюсти;
- искривление позвоночника; ;
- деформация ног;
- глухота;
- низкий уровень умственного развития.
Ребенок с таким генетическим заболеванием испытывает проблемы в езде на велосипеде, а также любом транспорте, который требует держания равновесия. Такие дети неуклюжи, они не могут ловить мелкие предметы. В 80-85% случаев отмечается интеллектуальная инвалидность.
В психоэмоциональном ракурсе присутствует раздражительность, чрезмерная возбудимость, быстрые перепады настроения, замкнутость и необщительность, очень редко немотивированная агрессивность.
Диагностика
Чтобы выявить общий характер отклонений, одного внешнего осмотра пациента будет недостаточно.

Синдром Сотоса требует тщательного обследования, включающего в себя:
- внешний осмотр;
- опрос родителей;
- проведение ультразвуковой диагностики внутренних органов на предмет нарушения;
- рентгенологическое исследование скелета, после чего отчетливо определяется опережение костного развития над физически, также может быть обнаружено искривление позвоночного столба с деформацией отдельных костей;
- компьютерную томографию головы, сердца, что помогает установить нарушения в структуре органа;
- магнитно-резонансную томографию головы, с ее помощью подтверждается недоразвитость мозолистого тела с отсутствием анатомических изменений;
- электроэнцефалографию, после нее видна сниженная активность нервной системы, присутствуют ЭЭГ-признаки нарушения взаимоотношения между корой и подкорковыми структурами;
- молекулярно-генетические процедуры – позволяют выявить ген с мутационными изменениями.
При выяснении психоневрологического состояния назначается консультация психиатра и невролога.
К дополнительным процедурам относятся:
- установление уровня гормонов гипофиза и надпочечников в крови;
- кардиологические обследования – помогут выявить наличие врожденного порока сердца.
После уточнения диагноза и выяснения всей картины нарушений назначаются соответствующие терапевтические методики.
Лечение
Синдром Сотоса в большинстве случаев имеет систематическое лечение: проводится коррекция деформаций во внутренних органах с помощью хирургических процедур.
При искривлении позвоночника и выраженной деформации костей применяются ортопедические методики.
Особо эффективны следующие мероприятия для коррекции отклонений в развитии:
- применение физиотерапевтических процедур с целью поддержания мышечного тонуса;
- поведенческая терапия используется при агрессивно настроенной личности и направлена на социализацию и улучшение коммуникативных особенностей;
- пациент находится на постоянном контроле у офтальмолога и стоматолога, чтобы вовремя принять меры по устранению изменений.
Дети с таким диагнозом требуют специального ухода и обучения. Прогноз для жизни положительный. Если вовремя оказана помощь таким детям, то они с течением времени смогут за собой ухаживать и выполнять все необходимые гигиенические процедуры и не только.
Возможные осложнения и профилактика
Синдром Сотоса опасен усугублением состояния пациента из-за порока сердца. У детей существует большой риск развития онкологических патологий. Если у человека проявляется излишняя агрессивность, то его госпитализируют в специальное медицинское учреждение с назначением успокоительных средств.
На данный момент единственным способом профилактики для родителей с таким диагнозом является выбор альтернативного оплодотворения или усыновление (удочерение) ребенка.
Читайте также:
- Чреспищеводная эхокардиография (ЧПЭ) через трансгастральный доступ
- Слизистая при эндофитном раке желудка. Рентгенография при раке желудка
- Связки укрепляющие коленный сустав. Связки коленного сустава. Коллатеральные связки коленного сустава. Внутрисуставные связки коленного сустава.
- Анаболические стероиды
- Мастит