Синдром Вискотта-Олдрича
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Под синдромом Вискотта-Олдрича понимают комплексную аутоиммунную патологию, характеризующуюся медленным образованием тромбоцитов. Медицинская литература содержит описания этой болезни, именуя её дефицитом иммунитета, сопровождающегося тромбоцитопениями и кожными проявлениями в виде экземы.
Чаще всего синдром Вискотта-Олдрича встречается у лиц мужского пола. Один из ведущих симптомов, который показывают анализы крови, — это тромбоцитопения, иными словами, сниженная скорость образования тромбоцитов. Их число в крови резко уменьшается, а размеры становятся недостаточными для осуществления полноценной функции свёртываемости крови.
Такую тромбоцитарную аномалию обычно наблюдают у людей уже с рождения. При осмотре кожа новорождённого ребёнка выглядит «смятой», а впоследствии после небольших травм возникают серьёзные кровотечения.
Происхождение заболевания
Болезнь имеет генетическое происхождение. Её вызывают генные мутации. Как правило, речь идёт о Х-хромосоме и её определённом участке (Хр 11 22-23). Мальчики, рождённые от женщины, носящей такой вид мутации, имеют высокую вероятность развития синдрома Вискотта-Олдрича, которая составляет около 50%. Что касается девочек, они чаще становятся носителями болезни.
Когда новорождённые дети постепенно утрачивают материнские защитные белки-глобулины, они резко заболевают разными инфекциями. Генная мутация, в результате которой индивидуальная сопротивляемость организма слабеет, обуславливает возникновение у детей раннего возраста:
- отитов;
- воспалений лёгких;
- менингитов;
- гемофилий;
- болезней, вызываемых золотистым стафилококком;
- в тяжёлых случаях — сепсисов.
Особенно тяжёлая клиническая картина наблюдается в этих случаях при вирусных инфекциях. Обычный герпес часто чреват диффузным инфицированием слизистых оболочек разных органов, а ветрянка может дать осложнения, представляющие угрозу для жизни ребёнка. При присоединении грибковых заболеваний все они заканчиваются выраженным кандидозом кожи.
Сама болезнь протекает как тяжело, так и легко. Около 80% пациентов страдают экземой и иными системными заболеваниями. Иногда развиваются и более серьёзные виды осложнений в виде эритродермий или абсцессов. Характерна также аллергия на молочные продукты и другую пищу (чаще всего, на орехи).
Патогенез синдрома Вискотта-Олдрича
Заболевание приводит к патологической недостаточности функций лейкоцитов, что негативным образом влияет на состояние иммунной системы. Лейкоциты либо плохо работают, либо полностью утрачивают свои иммуносохраняющие способности. Это приводит к высокому риску возникновения расстройств иммунного и воспалительного характера. Люди, имеющие в анамнезе синдром Вискотта-Олдрича, часто болеют:
- экземой;
- другими воспалениями кожных покровов;
- всеми видами инфекционных патологий.
Аутоиммунные нарушения, неизбежно возникающие при заболевании, заключаются в том, что организм начинает «атаковать» свои же ткани, органы и клетки. Существует большой риск возникновения злокачественных патологий в виде лимфом по типу Ходжкина и других.
Дефицит иммуноглобулина М — яркий симптом, характерный для СВО. Он характеризуется стойким уровнем тромбоцитопении в крови и иммунодефицитом гуморальной системы организма.
Бесплатная консультация по вопросам обучения
Наши консультанты всегда готовы рассказать о всех деталях!
Клиническая картина
Клиническая симптоматика болезни отличается разнообразием и, кроме экземы, может проявляться такими признаками:
- тромбоцитопенической пурпурой;
- импетиго;
- фурункулёзом;
- абсцессами тканей.
Кожа больного выглядит слишком плотной, приобретает глянцевый оттенок с бордовым или красноватым отливом. На некоторых её участках отмечаются шершавость, болезненность и чрезмерная жёсткость.
Другие органы тоже могут давать болезненную симптоматику:
- патологии барабанных перепонок;
- гаймориты, фронтиты;
- инфекционные поражения слизистых;
- фарингиты, ларингиты;
- кандидоз;
- увеличение селезёнки и печени;
- бронхиальная астма, бронхиты, пневмонии;
- увеличения лимфатических узлов.
Иногда присоединяется неврологическая симптоматика в виде внутричерепных кровотечений, бессонницы, дезориентации в окружающем пространстве.

Первый год жизни ребёнка, страдающего синдромом Вискотта-Олдрича, характеризуется классической триадой проявлений, на основании которой медики делают вывод о первичном диагнозе. Это кровотечения, кожная экзема и присоединение любого инфекционного заболевания, как правило, протекающего долго и тяжело.
Сложность заключается в том, что маленькие дети, которым ещё не исполнилось года, не могут объяснить, где у них болит. Врачам следует проявлять настороженность при появлении у детей кровавой диареи, внезапных синяков и петехий. Тревогу должны вызывать и грибковые поражения слизистой оболочки рта и на половых органах.
Иногда классическая триада синдрома Вискотта-Олдрича может отсутствовать. При этом, у детей наблюдают только один либо несколько симптомов в виде:
- стойких тромбоцитопений;
- других гематологических патологий;
- инфекций;
- экземы;
- аутоиммунных нарушений;
- злокачественных лимфом.
Пупочная ранка у детей после родов долго мокнет, кровоточит и не заживает. Что касается экземы, она может развиваться в умеренной либо тяжёлой форме. Принести больному облегчение можно, смазывая его кожу увлажняющими кремами либо кремами, содержащими стероидные гормоны.
Методы терапии
Лечение в основном является симптоматическим. Аспирин и иные НПВС назначать больным нельзя, так как они будут препятствовать работе и без того ослабленных тромбоцитов. Маленьким детям рекомендуется ношение защитного шлема, чтобы избежать серьёзных травм. Если у пациента наблюдается низкое содержание тромбоцитов в крови, облегчить состояние поможет переливание крови. Также больным иногда удаляют селезёнку и вводят внутривенно иммуноглобулины. При возникновении анемии её купируют введением железосодержащих лекарственных средств.
Синдром Вискотта-Олдрича — болезнь с серьёзным нарушением функции кроветворения. Один из непростых, но действенных методов лечения заключается в трансплантации стволовых клеток. Его осуществляют, переливая больному кровь из пуповины либо пересаживая ему костный мозг.
Вискотта-Олдрича синдром
Синдром Вискотта-Олдрича (СВО) – редкое наследственное заболевание, которое характеризуется сочетанием трех симптомов: кожной экземы, тромбоцитопении (то есть низкого уровня тромбоцитов) и иммунодефицита. Таким образом, СВО относится к первичным иммунодефицитам. Кроме того, у больных СВО значительно повышен риск развития злокачественных опухолей и аутоиммунных нарушений.
Частота встречаемости и факторы риска
СВО – редкое заболевание; частота его оценивается как 4-10 случаев на 1 миллион живых новорожденных. Встречается во всех регионах.
Так как СВО характеризуется Х-сцепленным наследованием, болеют почти исключительно мальчики (описаны лишь единичные случаи, где атипичная форма этого синдрома предполагается у девочек). Заболевание может быть унаследовано с вероятностью 50% в случае, если мать мальчика является носительницей дефектного гена; при этом сама она клинически здорова. Семьям, где уже были случаи СВО у детей, рекомендована консультация генетика.
Признаки и симптомы
Симптомы СВО обычно начинают проявляться уже в течение первых месяцев жизни. С возрастом они, как правило, прогрессируют.
Тромбоциты при СВО имеют уменьшенный размер, а их функция нарушена. Эти аномальные тромбоциты разрушаются в селезенке; в результате уровень тромбоцитов снижается, а размер селезенки может увеличиваться. Тромбоцитопения проявляется возникновением синяков, подкожных кровоизлияний (петехий) и кровотечений – например, нередки кровавый понос, кровотечения из носа и десен. Из-за этого у больного может также развиться анемия.
В большинстве случаев одним из симптомов является зудящая кожная сыпь, хотя у некоторых больных она может быть слабо выраженной или даже отсутствовать.
Иммунодефицит связан как с нарушением функции В-лимфоцитов, так и с дефицитом Т-лимфоцитов, то есть речь идет о комбинированном иммунодефиците. Его следствием являются упорные, часто повторяющиеся бактериальные, вирусные и грибковые инфекции, которые могут поражать верхние и нижние дыхательные пути, пазухи носа, уши, глаза, кожу, слизистые оболочки, желудочно-кишечный тракт, мочевыводящие пути.
У больных СВО существенно повышен риск возникновения злокачественных заболеваний, прежде всего лимфом и лейкозов; с возрастом этот риск увеличивается. В ряде случаев развиваются и аутоиммунные нарушения, такие как васкулит (поражение кровеносных сосудов) или гемолитическая анемия.
Конкретные генетические дефекты (мутации), вызывающие СВО, могут различаться у разных детей. Соответственно, тяжесть протекания болезни зависит от того, каким именно дефектом она вызвана в каждом случае. Все эти дефекты касаются гена особого белка WASP (Wiskott-Aldrich syndrome protein), который выполняет определенные функции в клетках крови. Если в результате мутации выработка этого белка почти полностью прекращается, то наблюдаются «классические», более тяжелые формы болезни; если же возможна выработка некоторых количеств измененного белка, то болезнь протекает не так тяжело.
Диагностика
СВО диагностируется на основе клинических симптомов, результатов клинического анализа крови, микроскопического исследования мазка крови (которое позволяет обнаружить, наряду с недостаточным уровнем тромбоцитов, их уменьшенный размер), а также измерения уровней различных иммуноглобулинов крови, которые изменены по сравнению с нормальными. Существуют и другие иммунологические методы, используемые при диагностике.
Точное подтверждение диагноза может быть произведено на основе либо обнаружения мутации в соответствующем гене на Х-хромосоме больного, либо измерения уровня белка WASP (см. выше) в клетках крови. Существенное снижение уровня этого белка указывает на синдром Вискотта-Олдрича.
Разработаны методы пренатальной, то есть до рождения ребенка, диагностики СВО.
Лечение
Лекарственная терапия не может привести к излечению СВО, но некоторые меры приводят к увеличению продолжительности и повышению качества жизни больных. Так, поскольку одной из основных проблем при СВО является иммунодефицит, для лечения инфекционных осложнений необходимо использовать соответствующие препараты – антибиотики и т.д. Возможно внутривенное введение иммуноглобулинов для коррекции их уровня в организме.
Для облегчения состояния больного могут применяться переливания компонентов крови, прежде всего тромбоцитов.
Так как тромбоциты разрушаются в селезенке, то иногда удаление селезенки (спленэктомия) может привести к улучшению состояния больного. Однако дети с удаленной селезенкой еще сильнее подвержены инфекциям.
Кожная сыпь требует местного лечения и тщательного ухода за кожей.
Больные СВО должны соблюдать ряд ограничений. Так, необходимы различные меры для профилактики инфекций. Противопоказаны прививки живыми вакцинами. Не следует давать ребенку аспирин и некоторые другие противовоспалительные средства, так как они влияют на свертываемость крови. Нужно избегать травм, которые могут вызвать кровотечения и кровоизлияния, а при очень низком уровне тромбоцитов детям младшего возраста иногда даже рекомендуют носить на голове защитный шлем – во избежание кровоизлияния в мозг при падениях.
Генетический дефект, вызывающий СВО, нарушает нормальное функционирование клеток кроветворной системы – именно с этим и связаны проявления болезни. Соответственно, аллогенная трансплантация костного мозга в случае успеха приводит к выздоровлению, поскольку кроветворные клетки больного заменяются донорскими клетками с нормальной копией гена. Именно трансплантация, несмотря на связанные с ней серьезные риски, является фактически единственным излечивающим методом при СВО. К сожалению, у большинства больных нет совместимого родственного донора, но в последние годы все чаще возможны успешные пересадки от неродственных или частично совместимых доноров. Решение о трансплантации и ее сроках принимает врач в зависимости от состояния больного, наличия донора и других факторов. Изучается применение при трансплантациях по поводу СВО препарата плериксафор (“Мозобаил”); есть данные, что он существенно улучшает приживление и повышает шансы на полное восстановление именно при СВО.
Прогноз
Без лечения прогноз при СВО плохой: большинство пациентов погибает в раннем детском возрасте от кровотечений или инфекций. Сейчас при использовании адекватной поддерживающей терапии (иммуноглобулины, антибиотики, компоненты крови) и соблюдении необходимых ограничений больные могут доживать до подросткового или даже взрослого возраста, хотя опасность развития тяжелых осложнений все время сохраняется.
Проведение трансплантации костного мозга существенно улучшает прогноз у многих больных СВО. Большинство трансплантаций от полностью совместимого родственного донора заканчивается успешно. Результаты трансплантаций от совместимых неродственных доноров и частично совместимых родственных доноров также постоянно улучшаются.
На Западе известны пациенты, получившие трансплантацию по поводу СВО 30 и более лет назад; сейчас они ведут нормальную полноценную жизнь.
Ученые МГУ раскрыли молекулярный механизм редкой генетической болезни
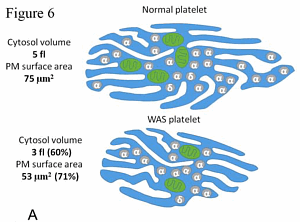
При синдроме Викскотта-Олдрича площадь поверхность и объем тромбоцитов непропорционально меньше, чем у здоровых тромбоцитов // Источник: S. Obydennyi, E. Artemenko, A. Sveshnikova, Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldric
Коллектив российских ученых при ведущей роли сотрудников МГУ изучил молекулярные механизмы, происходящие в клетках крови у детей с синдромом Вискотта-Олдрича. Оказалось, что при этой болезни в тромбоцитах нарушается отношение объема клетки к площади её поверхности, что приводит к нарушению кальциевого гомеостаза и запуску редкого механизма программированной гибели клетки — митохондриального некроза. Работа была поддержана фондом «Врачи, инновации, наука — детям», Российским фондом фундаментальных исследований (РФФИ) и Российским научным фондом (РНФ). Результаты исследования опубликованы в престижном журнале Haematologica (IF=7,6).
Синдром Вискотта-Олдрича — редкое генетическое заболевание, которое встречается в 1-10 случаях на 1 миллион человек по всему миру. Чаще оно проявляется у мужчин, поскольку вызвано мутациями гена WAS, находящимся в X-хромосоме. Этот ген кодирует белок WASP, который присутствует во всех типах клеток крови и отвечает за регуляцию актинового цитоскелета. Кровяные тельца с нарушенной работой белка WASP хуже распознают внешние факторы. Лейкоциты с поломкой в WASP не способны формировать адекватный иммунный ответ, а тромбоциты развиваются некорректно и склонны преждевременно умирать.
Почему при синдроме Вискотта-Олдрича запускается каскад реакций, приводящих к гибели клетки, науке было неясно. Коллаборация российских ученых выяснила молекулярные механизмы, протекающие в нарушенных тромбоцитах. «В этой работе мы обнаружили механизм запрограммированной клеточной смерти тромбоцитов при синдроме Вискотта-Олдрича, наследственной болезни, при которой дети умирают от кровотечений, — рассказал ведущий автор исследования, заведующий лабораторией клеточного гемостаза НМИЦ ДГОИ имени Дмитрия Рогачева и профессор кафедры медицинской физики физического факультета МГУ Михаил Пантелеев. — Этот механизм связан с нарушением соотношения объема/поверхность тромбоцита, которое ведет к нарушению кальциевого гомеостаза и гибели по пути митохондриального некроза».
Ученые в реальном времени следили за уровнем ионов Ca2+ в цитоплазме нарушенных тромбоцитов, за электрическим потенциалом их мембраны и изменениями сигнальных молекул фосфатидилсерина на ней. В норме молекулы фосфатидилсерина располагаются на внутренней поверхности мембран, однако при запуске каскада реакций программируемой клеточной гибели они перемещаются на внешнюю. Оказалось, что уровень ионов кальция в поврежденных тромбоцитах в состоянии покоя значительно выше, чем у нормальных клеток, поляризация и реполяризация мембраны проходят значимо чаще, а их митохондрии теряют потенциал с последующей экспозицией фосфатидилсерина после разрушения последней митохондрии.
Запуск механизма программируемой клеточной гибели в нарушенных клетках ученым удалось предотвратить путем удаления излишков ионов кальция из клеточного окружения и введения внутрь клетки блокаторов пор митохондриальной мембраны: циклоспорина А или квестоспонгина C. Введение тапсигаргина — вещества, которое не дает клетке откачивать ионы кальция во внутриклеточные депо из цитоплазмы, — наоборот, приводило к скорейшей гибели клетки. Функционирование тромбоцитов также зависело от числа митохондрий в них: чем их меньше, тем скорее запускались механизмы апоптоза.
На основе полученных данных ученые построили компьютерную модель. Модель показала, что «благополучие» тромбоцитов напрямую зависит от кальциевого гомеостаза: его нарушение приводит к экспонированию фосфатидилсерина на поверхность митохондриальной мембраны и гибели клетки по пути митохондриального некроза. Нарушение кальциевого гомеостаза, в свою очередь, происходит из-за нарушения соотношения площади поверхности к объему клетки. «Найденный механизм объясняет почему у детей с синдромом Вискотта-Олдрича мало тромбоцитов, что позволит предложить новые способы их лечения. Кроме того, эти данные проливают свет на механизмы клеточной смерти тромбоцитов даже в здоровых клетках, что пригодится при диагностике и лечении тромбозов и кровотечений», — заключил Михаил Пантелеев.
Заняться изучением синдрома Вискотта-Олдрича ученым предложили специалисты из Центра детской гематологии имени Дмитрия Рогачева, где лечатся дети с этим заболеванием, и они же предоставили генетически подтвержденные образцы крови пациентов с этим заболеванием. Помимо исследователей из МГУ имени М.В.Ломоносова и НМИЦ ДГОИ имени Дмитрия Рогачева, в работе приняли участие сотрудники Центра теоретических проблем физико-химической фармакологии РАН, Первого МГМУ имени И.М. Сеченова, Института эволюционной физиологии и биохимии имени И.М. Сеченова РАН, МГТУ имени Н.Э. Баумана и МФТИ.
Синдром Вискотта-Олдрича
Синдром Вискотта – Олдрича обусловлен комбинированной недостаточностью В- и Т-лимфоцитов и характеризуется рецидивирующими инфекциями, экземой и тромбоцитопенией.
Является сцепленным с Х-хромосомой наследственным заболеванием. Причиной развития синдрома Вискотта – Олдрича являются мутации гена, который кодирует белок синдрома Вискотта – Олдрича (БСВО, WASP – Wiskott-Aldrich syndrome protein), цитоплазматический протеин, необходимый для нормального обмена сигналами между Т- и В-лимфоцитами.
В связи с нарушением функционирования Т- и В-лимфоцитов у пациентов развиваются инфекции, вызванные гноеродными бактериями и оппортунистическими организмами, особенно вирусами и Pneumocystis jirovecii. Частыми являются инфекции, вызванные вирусом ветряной оспы Ветряная оспа Ветряная оспа – острая системная, обычно детская инфекция, вызываемая вирусом ветряной оспы – вирусом опоясывающего лишая (герпесвирус человека тип 3). Обычно начинается с умеренных общеинтоксикационных. Прочитайте дополнительные сведения .
Симптомы и признаки синдрома Вискотта-Олдрича
Первыми проявлениями часто являются геморрагические (обычно кровавый понос), а затем рецидивирующие инфекции дыхательных путей, экзема и тромбоцитопения.
Опухоли, особенно вирусные В-клеточные лимфомы (ВЭБ+) и острый лимфобластный лейкоз Острый лимфобластный лейкоз (ОЛЛ) Острый лимфобластный лейкоз (ОЛЛ) является наиболее частым онкологическим заболеванием у детей, но поражает также и взрослых всех возрастов. Злокачественная трансформация и неконтролируемая. Прочитайте дополнительные сведения 10 лет.
Диагностика синдрома Вискотта-Олдрича
Определение количества и объема тромбоцитов
Исследование функции лейкоцитов (например, хемотаксис нейтрофилов, функция Т-клеток)
Диагноз синдрома Вискотта-Олдрича основан на следующих критериях:
Снижение количества Т-клеток и функции
Повышенные уровни IgE и IgA
Низкие уровни IgМ
Низкие или нормальные уровни IgG
Снижение цитотоксичности естественных киллеров
Нарушенный хемотаксис нейтрофилов
Могут наблюдаться частичные дефекты антител к полисахаридным антигенам (например, к антигенам групп крови А и В). Тромбоциты маленькие, с дефектами, разрушение их в селезенке увеличено, что ведет к тромбоцитопении Обзор тромбоцитарных заболеваниях (Overview of Platelet Disorders) Тромбоциты – фрагменты клетки, которые функционируют в системе свертывания крови. Тромбопоэтин способствует контролю количества циркулирующих тромбоцитов, стимулируя костный мозг к выработке. Прочитайте дополнительные сведенияРекомендовано генетическое тестирование для родственников 1-й степени родства.
По причине увеличения риска развития лимфомы и лейкемии, обычно проводят общий анализ крови с подсчётом лейкоцитарной формулы каждые 6 месяцев. При наличии острых изменений в симптоматике, связанных с дисфункцией В-клеток, требуется более углубленная оценка.
Лечение cиндрома Вискотта-Олдрича
Поддерживающая терапия с профилактическим использованием иммуноглобулина, антибиотиков и ацикловира
При симптоматической тромбоцитопении применяют переливание тромбоцитарной массы и, в редких случаях, спленэктомию
Трансплантация гемопоэтических стволовых клеток
При лечении синдрома Вискотта-Олдрича для предотвращения рецидивирующих бактериальных инфекций применяют профилактическую терапию антибиотиками и иммуноглобулином Заместительная терапия недостающими иммунными компонентами Иммунодефициты обычно проявляются рецидивирующими инфекционными процессами. Однако причины при рецидивирующих инфекциях чаще всего отличаются от таковых при иммунодефиците (например, неадекватное. Прочитайте дополнительные сведения ; для профилактики серьезных вирусных инфекций простого герпеса используют ацикловир; при лечении геморрагии используют переливание тромбоцитарной массы. При тяжелой тромбоцитопении возможно применение спленэктомии, но обычно ее избегают по причине увеличения риска сепсиса.
Без трансплантации большинство пациентов умирают к 15 годам; однако некоторые пациенты могут дожить до зрелого возраста.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Особенности внутриклеточной кальциевой сигнализации тромбоцитов при синдроме Вискотта–Олдрича
Одна из ключевых особенностей синдрома Вискотта–Олдрича (СВО), редкого X-сцепленного иммунодефицитного состояния, – микротромбоцитопения, приводящая к спонтанным/посттравматическим кровотечениям. Причиной развития синдрома Вискотта–Олдрича является мутация в гене белка WASP, участвующего в поляризации актина и перестройке актинового цитоскелета. Механизм влияния данной мутации на внутриклеточную кальциевую сигнализацию, а также функциональные ответы тромбоцитов пациентов с СВО не уточнены. Цель исследования: анализ кальциевой сигнализации, изменения формы и связывания фибриногена тромбоцитами пациентов с СВО. Данное исследование поддержано Независимым этическим комитетом и утверждено решением Ученого совета НМИЦ ДГОИ им. Дмитрия Рогачева Минздрава России. В исследование были включены 3 пациента с СВО и 3 здоровых добровольца. Внутриклеточная сигнализация и функциональные ответы тромбоцитов наблюдались на проточном цитометре BD Facs Canto II. Для измерения концентрации кальция в цитозоле тромбоцитов использовали флуорофор Fura-Red; изменение формы тромбоцитов при активации оценивали по боковому светорассеянию клеток на длине волны 488 нм; активацию тромбоцитарных интегринов – по связыванию флуоресцентно-меченного фибриногена. Во время активации концентрация тромбоцитов составляла 1000 клеток/мкл во избежание эффекта вторичной активации. В покоящемся состоянии тромбоцитов наблюдалась повышенная концентрация кальция в цитозоле тромбоцитов пациентов по сравнению с тромбоцитами здоровых доноров. В ответ на стимуляцию максимально достижимые концентрации кальция были сопоставимы в обоих случаях. Связывание фибриногена с тромбоцитами пациентов не было значимо изменено по сравнению со здоровыми донорами. С другой стороны, изменение формы клеток в ответ на активацию, выраженное в процентах, у пациентов оказалось более значимым, чем изменение формы тромбоцитов здоровых доноров. При схожих максимальных ответах на стимуляцию всеми агонистами концентрация кальция в покоящихся тромбоцитах, а также изменение формы тромбоцитов у пациентов с СВО значимо выше, чем у тромбоцитов здоровых доноров. Данные результаты можно объяснить увеличенным отношением площади мембраны тромбоцитов к их объему.
Ключевые слова
Об авторах
ФГБУН «Центр теоретических проблем физико-химической фармакологии» РАН; ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России; ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова»; ФГБУН «Институт биохимической физики им. Н.М. Эмануэля» РАН
Россия
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
ФГБУН «Центр теоретических проблем физико-химической фармакологии» РАН; ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России; ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова»; ФГБОУ ВО «Московский физико-технический институт»
Россия
д-р физ.-мат. наук, профессор, зав. лабораторией клеточного гемостаза и тромбоза,
117997, Москва, ГСП-7, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
ФГБУН «Центр теоретических проблем физико-химической фармакологии» РАН; ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России; ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова»; ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России
Россия
Список литературы
1. Candotti F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott–Aldrich Syndrome. J Clin Immunol 2018; 38 (1): 13–27.
2. Perry G.S. 3rd, Spector B.D., Schuman L.M., Mandel J.S., Anderson V.E., McHugh R.B., et al. The Wiskott–Aldrich syndrome in the United States and Canada (1892- 1979). J Pediatr 1980; 97: 72–8.
3. Moratto D., Giliani S., Notarangelo L.D., Mazza C., Mazzolari E., Notarangelo L.D. The Wiskott–Aldrich syndrome: from genotype–phenotype correlation to treatment. Expert Rev. Clin Immunol 2007; 3: 813–24.
4. Rivers E., Thrasher A.J. Wiskott–Aldrich syndrome protein: Emerging mechanisms in immunity. Eur J Immunol 2017; 47: 1857–66.
5. Taylor M.D., Sadhukhan S., Kottangada P., Ramgopal A., Sarkar K., D’Silva S., et al. Nuclear Role of WASp in the Pathogenesis of Dysregulated TH1 Immunity in Human Wiskott–Aldrich Syndrome. Sci Transl Med 2010; 2: 37ra44 LP-37ra44.
6. Patel P.D., Samanich J.M., Mitchell W.B., Manwani D. A unique presentation of Wiskott–Aldrich syndrome in relation to platelet size. Pediatr. Blood Cancer 2011; 56: 1127–9.
7. Ochs H.D., Slichter S.J., Harker L.A., Von Behrens W.E., Clark R.A., Wedgwood R.J. The Wiskott–Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood 1980; 55: 243–52.
8. Haddad E., Cramer E., Riviere C., Rameau P., Louache F., Guichard J., et al. The thrombocytopenia of Wiskott Aldrich syndrome is not related to a defect in proplatelet formation. Blood 1999; 94: 509–18.
9. Prislovsky A., Zeng X., Sokolic R.A., Garabedian E.N., Anur P., Candotti F., Strom T.S. Platelets from WAS patients show an increased susceptibility to ex vivo phagocytosis. Platelets 2013; 24: 288–96.
10. Shcherbina A., Rosen F.S., Remold-O’Donnell E. Pathological events in platelets of Wiskott–Aldrich syndrome patients. Br J Haematol 1999; 106: 875–83.
11. Mahlaoui N., Pellier I., Mignot C., Jais J.-P., Bilhou-Nabera C., Moshous D., et al. Characteristics and outcome of early-onset, severe forms of Wiskott–Aldrich syndrome. Blood 2013; 121: 1510–6.
12. Sandrock K., Zieger B. Current Strategies in Diagnosis of Inherited Storage Pool Defects. Transfus Med Hemother 2010; 37: 248–58.
13. Ignatova A.A., Ponomarenko E.A., Polokhov D.M., Suntsova E.V., Zharkov P.A., Fedorova D.V., et al. Flow cytometry for pediatric platelets. Platelets 2019; 30 (4): 428–37.
14. Obydennyi S.I., Artemenko E.O., Sveshnikova A.N., Ignatova A.A., Varlamova T.V., Gambaryan S., et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott–Aldrich syndrome platelets. Haematologica 2019. DOI: 10.3324/haematol.2018.214460 [Epub ahead of print].
15. Medina S.S., Siqueira L.H., Colella M.P., Yamaguti-Hayakawa G.G., Duarte B.K.L., Dos Santos Vilela M.M., Ozelo M.C. Intermittent low platelet counts hampering diagnosis of X-linked thrombocytopenia in children: report of two unrelated cases and a novel mutation in the gene coding for the Wiskott–Aldrich syndrome protein. BMC Pediatr 2017; 17: 151.
16. Sokolic R., Oden N., Candotti F. Assessment of Immature Platelet Fraction in the Diagnosis of Wiskott–Aldrich Syndrome. Front Pediatr 2015; 3: 49.
17. Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J Thromb Haemost 2016; 14: 1867–81.
18. Fischer A. Platelets are the Achilles’ heel of Wiskott–Aldrich syndrome. J Allergy Clin Immunol 2019; 144: 668–70.
19. Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014; 312: 729–38.
20. Chiang S. Screening for Wiskott–Aldrich syndrome by flow cytometry. J Allergy Clin Immunol 2018; 142: 333–5.
21. Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., et al. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J Thromb Haemost 2016; 14: 2045–57.
22. Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985; 260: 3440–50.
23. Schoenmakers T.J., Visser G.J., Flik G., Theuvenet A.P. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques 1992; 12: 870–9.
24. Heemskerk J.W.M., Hoyland J., Masont W.T., Sage S. Spiking in cytosolic calcium concentration in single fibrinogen-bound fura-2-loaded human platelets. Biochem J 1992; 283: 379–83.
25. Poulter N.S., Pollitt A.Y., Davies A., Malinova D., Nash G.B., Hannon M.J., et al. Platelet actin nodules are podosome-like structures dependent on Wiskott-Aldrich syndrome protein and ARP2/3 complex. Nat Commun 2015; 6: 7254.
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Читайте также:
- Гемангиома носа. Нейрофиброма и неврилеммома околоносовых пазух.
- Гидроцефалия
- Возбудители госпитальных инфекций. Возбудители внутрибольничных инфекций.
- Цефалоспорины
- Терапия антикоагулянтами при инфаркте миокарда ( оим, ОИМ ). Тромболитическая терапия при инфаркте миокарда ( оим, ОИМ ). Стрептокиназа. Урокиназа.