Сопутствующие болезни синдрома Беквита-Видеманна в отоларингологии
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Над статьей доктора Вавилова Артема Сергеевича работали литературный редактор Елизавета Цыганок , научный редактор Екатерина Шрёдер и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Крипторхизм (Cryptorchidism) — это врождённый или приобретённый порок развития, при котором одно или оба яичка отсутствуют в мошонке. Иногда болезнь называют «неопущение яичка».
В норме у доношенного мальчика яички опускаются в мошонку к моменту рождения. Если этого не произошло, необходимо пройти осмотр у уролога или хирурга, после чего врач определит дальнейшие действия.
Распространённость крипторхизма
Крипторхизм — это один из наиболее частых пороков развития у мальчиков. По результатам систематического обзора, у доношенных младенцев и/или с массой тела больше 2,5 кг крипторхизм встречается в 1–4,6 % случаях, у недоношенных и/или с массой тела меньше 2,5 кг — 1,1–45,3 %. Частота болезни также зависит от возраста. Например, среди доношенных мальчиков и/или с массой тела при рождении больше 2,5 кг:
- в 1 год болеют 1–1,5 % детей;
- 6 лет — 0–2,6 %;
- 11 лет — 0–6,6 %;
- 15 лет — 1,6–2,2 % [1].
В группе риска находятся:
- недоношенные мальчики;
- малыши, размер которых не соответствовал гестационному возрасту (сроку беременности);
- младенцы, чей вес при рождении был меньше 2,5 кг.
По результатам метаанилиза, который охватил больше 111 000 младенцев, курение при беременности увеличивает риск развития крипторхизма в 1,18 раз [32].
Обычно крипторхизм — это изолированная патология. Однако он может сочетаться с эндокринными нарушениями, генетическими синдромами и аномалиями строения тела, особенно если крипторхизм двухсторонний. Сопутствующие состояния включают:
Все вышеперечисленные патологии встречаются редко. Однако у мальчиков с крипторхизмом генетические изменения наблюдаются чаще, чем у мальчиков без него. Так, изменения присутствовали у 17 из 600 детей с крипторхизмом (2,8 %), в то время как у мальчиков без крипторхизма частота генетических нарушений составила 0,3 % [25].
Крипторхизм также может быть приобретённым, то есть яичко сначала находилось в мошонке, но потом перестало в ней определяться. Такой крипторхизм может возникнуть после операции на паховой грыже или травмы, когда яичко подтягивается кверху из-за формирования рубцовой ткани.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы крипторхизма
Основной симптом крипторхизма — это отсутствие одного или обоих яичек в мошонке. Также важно обращать внимание на состояние мошонки. Из-за отсутствия яичка она неправильно развивается: на её коже меньше складок, она менее эластичная. При пальпации яичко в мошонке не прощупывается.
Если в мошонке нет яичка, но обе её половины выглядят одинаково, это может говорить об избыточно подвижном яичке, которое поднимается вверх. Некоторые врачи называют это состояние ложным крипторхизмом, так как часто яичко опускается на место самостоятельно. Такая подвижность связана с высоким рефлексом мышцы-кремастера, которая поднимает яичко из мошонки в паховый канал.
При избыточно подвижных яичках нужно наблюдаться у хирурга или уролога примерно раз в 3–6 месяцев [3].
Патогенез крипторхизма
Механизмы, которые отвечают за нормальное опускание яичек, ещё недостаточно изучены. Дефицит гонадотропина в утробе матери, снижение антимюллерова фактора, который отвечает за формирование пола, и повышенная концентрация эстрадиола в плаценте — это предположительные факторы, нарушающие процесс опускания яичка.
При истинном крипторхизме яичко идёт по «правильному» пути в мошонку, но не доходит до неё.
Также к крипторхизму относится эктопия — это состояние, когда яичко отклоняется от нормального пути опущения и находится вне мошонки. Чаще яичко располагается в паховой области, но может определяться на бедре, в промежности или лобковой области. Эктопию яичка обычно лечат с помощью операции. Когда яичко не прощупывается, в 50–60 % случаев оно находится внутри брюшной полости, в паховой области или у входа в паховый канал. У 20 % мальчиков яичко отсутствует, в 30 % случаев оно атрофировано или недоразвито [31].
Если яичко находится внутри брюшной полости, чаще всего оно располагается рядом с внутренним паховым кольцом, но иногда яичко находят около почки, на передней брюшной стенке или позади мочевого пузыря.
Отсутствие яичка (монорхизм) диагностируют у 4 % мальчиков с неопущением яичка. Во внутриутробном периоде семенные канатики могут перекрутиться, из-за чего нарушается кровообращение, яичко атрофируется и после рождения уже не определяется.
Двухстороннее отсутствие яичка (анорхизм) встречается в менее 1 % случаев. Такое состояние связывают с перекрутом яичковых сосудов и последующим внутриутробным инфарктом яичка (омертвением тканей) [31].
Классификация и стадии развития крипторхизма
Наиболее простая классификация крипторхизма — это разделение на прощупываемые и непрощупываемые яички.
Приблизительно 80 % неопущенных яичек удаётся прощупать [2]. Их можно разделить:
- на истинно неопущенные яички — прощупываются в паховом канале или сразу за наружным паховым кольцом и находятся в этом положении с рождения;
- ретрактильные (избыточно подвижные) яички — находятся выше мошонки, но при осмотре могут опускаться до мошонки и оставаться там, пока их удерживают, однако после этого вновь поднимаются;
- эктопированные яички — могут располагаться в промежности, паховой, надлобковой области и в области бедра, т. е. яички изначально развивались правильно, но впоследствии оказались в другом месте.
При сочетании одностороннего непрощупываемого яичка и гипоспадии врач проверяет, нет ли у ребёнка нарушений полового развития. В этом случае также нужна консультация генетика или эндокринолога.
Непрощупываемое одностороннее яичко без порока развития уретры можно разделить:
- на истинное неопущенное яичко — определяется нормальное яичко и его сосуды на пути естественного следования;
- отсутствующее яичко — также отсутствуют все его структуры, но можно найти их остатки. Это возможно при нарушении внутриутробного кровообращения (например, внутриутробном перекруте яичка).
- эктопии яичка — нормальное яичко и его сосуды находят в неправильном месте [30].
По результатам ретроспективного анализа, яички не прощупывались у 24 % пациентов с неопустившимися яичками. Среди 447 случаев непрощупываемых яичек у 41 % пациентов гонады были атрофированы или отсутствовали, у 20 % располагались в брюшной полости, у 30 % — в паховом канале и у 9 % находились в других местах [26].
Осложнения крипторхизма
К потенциальным осложнениям истинного крипторхизма относят:
- паховую грыжу;
- повышенный риск травмы и перекрута яичка;
- ухудшение работы репродуктивной системы вплоть до бесплодия;
- злокачественные образования [27][30].
Диагностика крипторхизма
Крипторхизм может обнаружить врач-неонатолог при первичном осмотре ребёнка сразу после рождения или педиатр во время наблюдений новорождённого на дому после выписки из родильного дома. Чтобы подтвердить диагноз и определить форму болезни, необходимо проконсультироваться с хирургом или урологом.
Основным диагностическим методом является тщательный физикальный осмотр. Сначала врач осматривает нормальный путь опущения яичка. Если яичка там нет, доктор прощупывает область бедра, промежность и лобковую зону.
Детей до года обследуют в положении лёжа на спине, в более старшем возрасте (после 4 – 6 недель) — стоя и лёжа. При одностороннем непрощупываемом яичке врач осматривает противоположное яичко: оценивает его размер и положение. Если оно увеличено, это может говорить об отсутствии или атрофии второго яичка, но этот симптом не специфичный, поэтому врач продолжает поиск [4][5].
Если у ребёнка двухсторонний крипторхизм или непрощупываемые яички, а также есть другие признаки нарушения полового развития (например, гипоспадия), врач уточняет диагноз с помощью тщательного осмотра малого таза и брюшной полости. Мальчика также обследует генетик и эндокринолог. Если после мультидисциплинарного дообследования нарушение подтверждается, врачи принимают решение о дальнейшей тактике лечения.
Первоначальное обследование новорождённого с двусторонним крипторхизмом включает:
- УЗИ брюшной полости и таза — обычно проводят в первую очередь;
- определение кариотипа — анализ, для которого берут кровь из вены, назначают после консультации генетика;
- исследование гормонов и метаболитов надпочечников — помогает оценить врождённое увеличение коры надпочечников, проводится по показаниям врача;
- определение лютеинизирующего гормона (ЛГ), фолликулстимулирующего гормона (ФСГ), тестостерона и мюллерова ингибирующего фактора (МИФ) — эти исследования проводят в возрасте около 6 недель по показаниям врача [30];
- осмотр паховой области — помогает определить, где находится яичко, и обнаружить паховую грыжу, если она есть.
В более старшем возрасте, после периода новорождённости, диагноз также уточняют с помощью эндокринологического обследования: определяют уровень тестостерона и проводят тест стимуляции хорионического гонадотропина (ХГЧ), чтобы исключить вторичную атрофию яичек в результате дефицита тестостерона [24][27].
Как правило, окончательный диагноз устанавливают с помощью диагностической лапароскопии и обследования паховой области.
Помимо осмотра яичек, необходимо провести полное обследование гениталий: осмотреть половой член, мошонку и паховые области, оценить расположение наружного отверстия уретры (меатуса). Это необходимо, чтобы исключить другие нарушения и осложнения, которые могут сочетаться с крипторхизмом, например:
- маленький половой член — может быть признаком нарушения полового развития или гипопитуитаризма;
- неправильное расположение меатуса — чем отверстие уретры ближе к промежности, тем выше вероятность нарушений полового развития;
- недоразвитая или плохо покрытая складками мошонка — снижает вероятность самостоятельного опускания яичка;
- раздвоение мошонки — может быть признаком нарушения полового развития.
Чтобы найти непрощупываемое яичко, могут выполнить УЗИ, однако достоверность этого метода остаётся низкой. По результату систематического исследования, чувствительность и специфичность ультразвука при обнаружении непрощупываемых яичек составила 45 и 78 % соответственно [28].
УЗИ используют при дифференциальной диагностике или планировании хирургического вмешательства, например:
- УЗИ может быть показано для поиска гонад (яичек) или матки у мальчика младше 1 года с двухсторонним непрощупываемым яичком — помогает оценить нарушения полового развития;
- УЗИ могут порекомендовать мальчикам с ожирением, у которых трудно провести пальпацию, — в зависимости от результатов врач может сменить тактику лечения: провести открытую операцию вместо лапароскопии.
При крипторхизме не используют компьютерную томографию из-за высокой дозы облучения и МРТ из-за высокой стоимости исследования, которое зачастую требует седации или анестезии.
Если врач так и не выявил местоположение яичка, это не значит, что его нет. Часто яичко находят с помощью лапароскопии, поэтому она считается главным методом диагностики и может использоваться дальше при лечении [28].
Лечение крипторхизма
Так как опущение яичка зависит от гормонов, раньше при лечении врачи использовали ХГЧ или гонадотропин-рилизинг гормон, однако такая терапия имела лишь 20 % успеха [8]. Даже при успешном опущении яичка в 20 % случаев появлялся риск повторного подъёма [9]. Чем выше располагалось яичко до начала лечения, тем ниже вероятность успеха гормонотерапии [10]. Сейчас лечить крипторхизм с помощью гормонов не рекомендуют [31].
Если яичко не опускается в мошонку к 6-му месяцу, то вероятность его самоопущения крайне низкая, поэтому необходимо проводить операцию [6][11]. Операция по опущению яичка в мошонку называют орхипексией. Ранняя орхипексия может частично ускорить рост яичка, чего не наблюдается при поздней операции, поэтому оптимальным сроком для проведения хирургического вмешательства является возраст от 6 до 12 месяцев [12][13]. Лечение рекомендуют завершить к 12–18 месяцам, так как микроскопические исследования неопущенных гонад показывают, что уже в этом возрасте мальчики начинают терять половые клетки и клетки Лейдига, которые продуцируют гормоны [7]. Это говорит о том, что неопущенные яички могут оказать негативное влияние на дальнейшую работу репродуктивной системы.
Если не сделать операцию вовремя, повышается риск развития следующих осложнений:
- Паховой грыжи — в 60–90 % случаях крипторхизм сочетается с открытым влагалищным отростком (причиной паховой грыжи) [29].
- Травмы — риск травмы увеличивается при неопущенном яичке. Нормальное положение яичка позволяет ему двигаться при ударе, что невозможно при крипторхизме. Также прикрепление яичка к тканям позволяет избежать травм в результате сдавливания сокращёнными мышцами паха.
- Опухоли — при неопустившемся яичке риск развития опухоли повышается, особенно когда яичко находится в брюшной области.
- Орхоэпидидимита — воспаления яичка в придатке в результате травмы в паховом канале.
- Бесплодия — температура в паху и брюшной полости на 1,5–2 °С выше, чем в мошонке. Операция помогает предотвратить повреждение яичка, связанное с повышенной температурой. Чем дольше яичко находится в ненормальном положении, тем выше риск бесплодия.
- Перекрута — низведение яичка предотвращает его перекрут, вызванный неправильным прикреплением [29].
Операцию также проводят, исходя из эстетических показаний: орхипексия помогает восстановить привычный симметричный вид мошонки.
При прощупываемом яичке обычно проводят орхипексию с помощью рассечения мягких тканей паха, при этом её эффективность достигает 92 % [14]. При операции важно отделить яичко и семенной канатик до уровня внутреннего пахового кольца, после чего разрезать мышцу, которая поднимает яичко, и связку, направляющую его. Это предотвращает повторный выход яичка из мошонки. Его укладывают в «карман» из мясистой оболочки мошонки и там подшивают. Также необходимо тщательно выделить и рассечь влагалищный отросток брюшины на уровне внутреннего пахового кольца. Если этого не сделать, повышается риск рецидива [15].
Во время операции можно удалить отростки придатка яичка (гидатиду Морганьи), оценить объём и консистенцию яичка, а также связь яичка и придатка. Значительное расстояние между придатком и яичком может ухудшить прогноз фертильности (способности зачать ребёнка).
При непрощупываемых яичках цель операции — определить, имеется яичко или нет. После того как врач его обнаружил, он решает, удалять яичко или низводить в мошонку. Повторный осмотр под общей анестезией иногда помогает найти ранее непрощупываемое яичко.
Самым простым и точным способом обнаружить непальпируемое яичко остаётся диагностическая лапароскопия брюшной полости. С помощью камеры врач может найти:
- яичко в брюшной полости — 40 % случаев;
- яичко в паховом канале — 10 %;
- семенные сосуды, которые входят в паховый канал — 40 %;
- слепо заканчивающиеся семенные сосуды — 10 %.
Слепо заканчивающиеся сосуды говорят об исчезновении яичка. Они могут возникнуть при внутриутробном перекруте или ишемии яичка. После такой находки операция заканчивается [16].
Если сосуды входят в паховый канал, врач исследует паховую область. Когда он находит здоровое или атрофированное яичко, то следом проводит стандартную орхипексию.
Иногда яичко находится далеко от входа в паховый канал, поэтому низвести его в мошонку стандартным способом невозможно. Тогда используют операцию Фаулер — Стивенса, при которой разрезают яичковые сосуды. При этом кровь к яичку поступает через артерию семявыносящего протока и сосуды мышцы, поднимающей яичко.
После рассечения сосудов яичко опускают по направлению к мошонке. Как правило, полностью низвести яичко за одну операцию не получается, так как сосуды ещё недостаточно развиты: их длина не позволяет свободно опустить яичко, а если сосуды «перетянуть», кровоснабжение будет нарушено и яичко атрофируется. Повторную операцию проводят через 6 месяцев. Частота сохранения яичек при одноэтапной операции составляет 50–60 %, при двухэтапной эффективность повышается до 90 % [17].
Атрофия яичка — наиболее серьёзное осложнение хирургического лечения. По результатам систематического обзора, общая частота атрофии яичка при первичной орхипексии составляет 1,83 %, при одноэтапной операции Фаулера — Стивенса — 28,1 %, при двухэтапной — 8,2 % [18]. Среди осложнений после операции также выделяют повторное поднятие яичка, повреждение семявыносящего протока или самого яичка, раневую инфекцию и гематому.
Прогноз. Профилактика
Мужчины с неопущением одного яичка становятся отцами почти также часто, как и мужчины без крипторхизма. При двухстороннем крипторхизме уровень отцовства понижается.
Исследования, оценивающие гормональные изменения, доказали преимущество более раннего хирургического вмешательства при крипторхизме [19]. Также учёные обнаружили, что неопущение яичка связано с повышенной потерей половых клеток и клеток Лейдига. Поэтому, чтобы сохранить фертильность, важно своевременно лечить крипторхизм [20].
Двухстороннему крипторхизму в 100 % случаев сопутствует олигоозоспермия (снижение количества сперматозоидов), в 75 % — азооспермия (отсутствие сперматозоидов в семенной жидкости). После успешного лечения у 75 % пациентов сохранялась олигоозоспермия, у 42 % — азооспермия [21].
У больных с крипторхизмом повышен риск развития опухолей яичка, при этом риск понижается, если лечение провели в допубертантный период: у мужчин, которые перенесли операцию по поводу крипторхизма до 13 лет, опухоль развивается в 2,23 раза чаще, чем среди жителей в целом, а у больных, прошедших хирургическое вмешательство после 13 лет — в 5,4 раза [22]. У мальчиков с изолированным крипторхизмом в 3 раза больше шансов заболеть раком яичек [23].
Профилактика крипторхизма
Профилактики крипторхизма нет, но важно соблюдать врачебные рекомендации после лечения и возвращения яичка не место и проходить необходимые осмотры:
- через неделю после операции — врач оценивает заживление послеоперационных ран;
- через 3 месяца — контрольный осмотр, при котором доктор проверяет, атрофировалось ли яичко и где оно расположено;
- в начале полового созревания (13–15 лет) и после его окончания (18–19 лет) — иногда необходимо проверить уровень тестостерона и выполнить спермограмму.
Чтобы предупредить развитие злокачественного образования, важно иногда проводить самообследование. Уточнить, как правильно это делать, можно у лечащего врача.
Сопутствующие болезни синдрома Беквита-Видеманна в отоларингологии
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова;
Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Центр лечения бесплодия "ЭКО", Москва
Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай)
Журнал: Проблемы репродукции. 2014;(3): 58‑61
Назаренко Т.А., Зыряева Н.А. Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай). Проблемы репродукции. 2014;(3):58‑61.
Nazarenko TA, Zyriaeva NA. The birth of the child with Beckwith-Wiedemann syndrome after IVF/ICSI (a case report). Russian Journal of Human Reproduction. 2014;(3):58‑61. (In Russ.).
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова;
Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Представлено клиническое наблюдение - рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ.
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова;
Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Центр лечения бесплодия "ЭКО", Москва
В основе импринтинга лежат различия в экспрессии генов, обусловленные метилированием ДНК. У человека предполагают наличие 300-500 импринтированных генов. В основном они кодируют ростовые факторы, одни экспрессируют с отцовской, другие - с материнской хромосомой 5. Клетки каждого человека содержат отпечатки (характер метилирования) от обоих родителей. При гаметогенезе эти отпечатки стираются и устанавливается характер метилирования, присущий женскому или мужскому организму. При оплодотворении эмбрион получает по одному набору от каждого родителя с соответствующим характером экспрессии. В случае попадания в процессе оплодотворения двух гомологичных хромосом от одного родителя это состояние называется однородительской дисомией (ОРД) [5, 6].
Нарушения метилирования возможны при гаметогенезе, в процессе оплодотворения и преимплантационного развития. Нарушения метилирования могут происходить на разных уровнях организации генома (геномный, хромосомный, генный), вызывая соответствующие отклонения в развитии или болезни импринтинга [4, 6, 7]. Наиболее распространены и изучены у человека синдромы Прадера-Вили, Ангельмана, Видемана-Беквита, Сильвера-Рассела и некоторые другие.
Синдром Видемана-Беквита, или EMG-синдром по первым буквам характерной триады - омфалоцеле, макроглоссия, гигантизм, впервые описан патологом J. Beckwith (США) в 1963 г. и педиатром H.-R. Wiedemann (Германия) в 1964 г. Синдром встречается с частотой 1 случай на 10-12 тыс. новорожденных, при этом 85% - спорадические случаи и только 15% - наследственные (аутосомно-доминантный тип наследования с неполной пенетрантностью, передача по материнской линии) [8].
Критерии диагностики в настоящее время четко не установлены, но диагноз ставится при наличии 3 больших и 1 малого диагностического критерия. Из больших критериев наиболее характерны макросомия, макроглоссия, омфалоцеле, аномалии почек, эмбриональные опухоли. Из малых критериев характерны многоводие, неонатальная гипогликемия. Дифференциальный диагноз проводится со следующими синдромами: синдромом Симпсона-Голаби-Бехмеля (макросомия, висцеромегалия, макроглоссия, аномалии почек; Х-сцепленный рецессивный тип наследования), синдромом Перлмана, синдромом Костелло, синдромом Сотоса, синдромом Марото-Лами (мукополисахаридоз IV типа) [8].
Молекулярной основой синдрома Видемана-Беквита являются нарушения в генах короткого плеча хромосомы 11. Участок на коротком плече 11-й хромосомы включает две группы импринтированных генов - домен 1 (гены ИПФР2, Н19 и ЦИ1) и домен 2 (гены CDKN1C, KCNQ1 и ЦИ2) (ЦИ - центр импринтинга) [8].
Этиология синдрома сложна и включает 5 групп нарушений. Нарушения метилирования 1 и 2 ЦИ (потеря метилирования ЦИ2 материнского аллеля CDKN1C - 50% и гиперметилирование ЦИ1 материнского аллеля ИПФР2 и Н19 - 5%), а также отцовская ОРД по 11-й хромосоме - 20% - встречаются наиболее часто. Нередки мутации гена CDKN1C материнского аллеля: спорадические - 5%, наследственные - 40%. Также встречаются дупликации, инверсии, транслокации в этих доменах - 1% и субмикроскопические нарушения - частота не определена. В 20% случаев характер нарушений не известен [8].
Учитывая возможность нарушений метилирования в процессе гаметогенеза, оплодотворения и преимплантационного развития, встает закономерный вопрос о влиянии вспомогательных репродуктивных технологий (ВРТ) на частоту возникновения болезней импринтинга. По данным литературы 9, многие авторы отмечают повышенный риск болезней импринтинга после применения ВРТ. Некоторые авторы [15, 16] не отмечают повышения риска. Другие связывают болезни импринтинга у потомства с исходными нарушениями у родителей при бесплодии [17]. Также имеются отдельные клинические наблюдения [18, 19].
Представляем клиническое наблюдение.
Пациентка И., 1980 года рождения, рост 176 см, масса 62 кг. Наследственность не отягощена. Из соматического анамнеза обращает на себя внимание наличие пролапса митрального клапана, гипотонии и с 12 лет брадикардии. В детстве состояла на учете по поводу хронического пиелонефрита, с учета снята: аппендэктомия в 16 лет; страдает хроническим колитом. Является носителем вируса простого герпеса (ВПГ1), периодически отмечает обострения лабиального герпеса. Менархе в 12 лет, менструальный цикл регулярный. Половая жизнь с 18 лет. Беременностей не было. Брак первый с 2008 г., муж - 1978 года рождения, здоров. Гинекологический анамнез отягощен оперативными вмешательствами: 2 раза было экстренное чревосечение по поводу разрыва эндометриоидных кист яичников (в 2004 и 2008 г.). В 2006 г. была проведена лапароскопия, разделение спаек в малом тазу и гистерорезектоскопия, рассечение неполной внутриматочной перегородки. В 2012 г. - гистероскопия, раздельное диагностическое выскабливание перед проведением повторного цикла ЭКО.
Женщина обратилась по поводу бесплодия в 2008 г. Диагноз: первичное бесплодие; наружный генитальный эндометриоз IV степени; спаечный процесс в малом тазу; седловидная матка; хронический эндометрит.
Проведено 3 цикла ЭКО/ИКСИ по длинному протоколу (агонисты ГнРГ + чМГ + рФСГ), при пункциях получено по 3 ооцита, проведен перенос 2 эмбрионов на 3-и сутки, беременность наступила в 3-м цикле, одноплодная.
Беременность протекала с угрозой прерывания в I триместре, с 23 нед выявлено многоводие, с 27 нед - двусторонний уретерогидронефроз у плода по данным УЗИ. В 34 нед беременности отмечено многоводие крайней степени выраженности по данным УЗИ, нарушение кровотока у плода по данным допплеровского исследования, произведено кесарево сечение в экстренном порядке, родился живой недоношенный мальчик массой тела 2850 г, длиной 46 см, оценка по шкале Апгар 3-7 баллов. Мама выписана на 10-е сутки, послеоперационый период протекал без осложнений.
Мальчик родился в октябре 2009 г. Диагноз основной: множественные врожденные пороки развития; порок центральной нервной системы (незрелость структур головного мозга); макросомия; макроглоссия; неонатальный сахарный диабет; правосторонний пузырно-мочеточниковый рефлюкс III-IV степени; дефект межжелудочковой перегородки. Осложнения: церебральная ишемия II степени; геморрагический синдром (легочное и желудочное кровотечение). Сопутствующий: тяжелая асфиксия при рождении; внутриутробная пневмония; анемия новорожденных; недоношенность 34 нед.
В родильном зале ребенку произведена интубация трахеи, переведен на искусственную вентиляцию легких. Ребенок наблюдался в течение 1 мес жизни в отделении интенсивной терапии новорожденных. Отмечались плотные распространенные отеки туловища, синдром угнетения ЦНС (снижен мышечный тонус и угнетены рефлексы), в последующем на фоне терапии отмечена положительная динамика. По поводу гипергликемии с рождения получал инсулинотерапию (актропид внутривенно капельно), был взят генетический анализ на неонатальный сахарный диабет. Был установлен уретральный катетер, затем наложена пункционная цистостома, после чего признаки дилатации чашечно-лоханочной системы и обоих мочеточников по данным УЗИ купированы.
По данным магнитно-резонансной томографии (МРТ) выявлено нарушение формирования структур головного мозга: картина наружной гидроцефалии, гипоплазии височных долей, нарушения процесса формирования борозд головного мозга, задержки миелинизации. При консультации генетиком на основании наличия синдромальной формы макросомии и множественных врожденных пороков развития поставлен диагноз: синдром Симпсона-Голаби-Бехмеля. Взят цитогенетический анализ. Кариотип 46XY, нормальный мужской.
В возрасте 1 мес ребенок переведен в ДГКБ №1. На первом году жизни произведены операции по поводу паховой грыжи, глоссомегалии. В 7 мес жизни на консультации генетика установлен диагноз: синдром Видемана-Беквита (макросомия, макроглоссия, задержка психомоторного развития - гипоплазия височных долей по данным МРТ). Ребенок наблюдался у невролога, челюстно-лицевого хирурга. В 2 года на консультации генетика поставлен диагноз: синдром Видемана-Беквита (макроглоссия, мегауретер с гидронефротической трансформацией. Аномалия головного мозга на МР-томограмме. Задержка темпов психомоторного развития. Гипертензионно-гидроцефальный синдром). Ребенок получал лечение у нефролога, невролога, проходил скрининг 4 раза в год по поводу возможных эмбриональных опухолей (УЗИ, альфа-фетопротеин крови).
Пациентка повторно обратилась с целью достижения беременности в августе 2012 г. Естественно возник вопрос о возможностях профилактики и диагностики синдрома Видемана-Беквита при повторной беременности у женщины. Пациентка была проконсультирована генетиками из Медико-генетического центра РАМН. К сожалению, в настоящее время точная лабораторная диагностика болезней импринтинга четко не разработана, поэтому молекулярно-генетическое обследование ребенка и супружеской пары не проводилось. При медико-генетическом консультировании синдром Видемана-Беквита у ребенка был расценен как спорадический, поэтому риск при повторной беременности был расценен как низкий.
Учитывая упомянутые молекулярные особенности, лежащие в основе болезней импринтинга, проведение преимплантационной генетической диагностики (ПГД) также не представлялось возможным.
Проведен 4-й цикл ЭКО - по длинному протоколу (агонисты ГнРГ, чМГ, рФСГ), при трансвагинальной пункции получено 2 ооцита, 27.09.12 перенесено 2 эмбриона 3-го дня развития, наступила вторая беременность (последняя менструация 09.09.12).
В I триместре у женщины был впервые выявлен первичный субклинический гипотиреоз, в течение всей беременности пациентка принимала эутирокс 75-50 мкг/сут. В остальном беременность протекала без особенностей.
По результатам 1-го и 2-го скринингов и УЗИ отклонений от нормы не отмечено. При медико-генетическом консультировании показаний для проведения инвазивной пренатальной диагностики не выявлено. Кроме того, в отношении болезней импринтинга возможности пренатальной диагностики также четко не установлены.
Согласно информации, полученной из источников в Интернете, методы лабораторной диагностики синдрома Видемана-Беквита, имеющиеся на современном этапе развития медицины, находятся на стадии разработки и лишь частично внедрены в практику, в основном в США, странах Европы. Методы лабораторной диагностики зависят от генетической этиологии и включают следующие способы: 1) анализ метилирования с помощью метилчувствительной множественной полимеразной цепной реакции (МЧ мПЦР; MS-MLPA); МЧ ПЦР; 2) анализ однородительской дисомии с помощью исследования однонуклеотидного полиморфизма, МЧ мПЦР (MS-MLPA) и ПЦР; 3) определение мутаций с помощью анализа последовательностей; 4) определение дупликаций, инверсий и транслокаций с помощью цитогенетического анализа и FISH и, возможно, современного метода сравнительной геномной гибридизации; 5) выявление субмикроскопических нарушений с помощью анализа микроделеций и микродупликаций [7, 8, 20, 21].
Тактика медико-генетического консультирования также зависит от генетической причины синдрома Видемана-Беквита, последняя определяет и степень риска для сибсов пробанда [8].
У 85% больных с синдромом Видемана-Беквита наследственность не отягощена и кариотип нормальный. При этом высокий риск (50%) существует при наличии мутаций гена CDKN1C и микроделеций, микродупликаций; в остальных случаях риск низкий. В этих случаях проводят определение мутаций у пробанда и родителей, а также в семье.
При невыясненной причине (мозаицизм при ОРД) риск эмпирически низкий.
У 10-15% больных наследственность отягощена и нормальный кариотип.
При выявлении мутации CDKN1C у пробанда (40%): при наличии мутации у одного из родителей риск составляет 50%, при отсутствии мутации у родителей риск низкий. Но возможен мозаицизм. При отсутствии мутации CDKN1C у пробанда (60%) риск для сибсов составляет до 50%.
Пренатальная диагностика проводится следующим образом [8].
В случае наличия больного ребенка в семье: при выявлении мутаций проводят амниоцентез и анализ ДНК. Теоретически возможен анализ метилирования в амниоцитах. При всех беременностях риска: в 16 нед при наличии омфалоцеле определяют альфа-фетопротеин крови, проводят УЗИ в 19-20 нед и 25-32 нед беременности для выявления пороков развития и макросомии.
При неотягощенной наследственности: в случае выявления изолированного омфалоцеле при УЗИ возможен амниоцентез, анализ метилирования и мутации гена СDKN1С, дупликации, инверсии, транслокации. Проводят динамическое УЗИ во время беременности для выявления пороков развития и макросомии.
У новорожденного проводят мониторинг неонатальной гипогликемии. Диагноз подтверждают вышеуказанными способами [8].
Вопрос о преимплантационной диагностике не решен. Теоретически возможно применение метода сравнительной геномной гибридизации в случае отягощенной наследственности и наличия мутаций в семье.
Сопутствующие болезни синдрома Беквита-Видеманна в отоларингологии
Сопутствующие болезни синдрома Беквита-Видеманна в отоларингологии
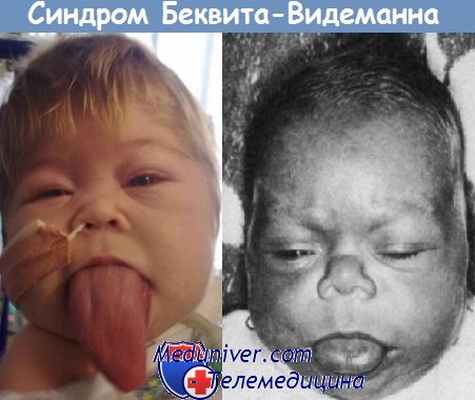
Синдром Беквита-Видеманна встречается с частотой 1:13700. Диагноз ставится клинически на основании наличия трех больших признаков или двух больших и одного малого признаков.
К большим признакам относятся: дефекты брюшной стенки, макроглоссия, макросомия, наличие борозд передней поверхности мочки или впадин в заушной области, висцеромегалия, омфалоцеле, опухоль Вильмса, гепатобластома, гемигиперплазия и специфические аномалии почек.
Многие признаки синдрома связаны с нарушением экспрессии генов, локализованных на хромосоме 11р 15.5; около 85% случаев вызваны спонтанной мутацией. Диагноз может быть заподозрен еще внутриутробно по ускоренным темпам развития плода во второй половине беременности. Новорожденные обычно входят в 97 процентиль по высоте и весу, и в 50-ый по размеру головы.
Внешний вид пациентов часто напоминает детей, чьи матери страдали гестационным диабетом (крупные для своего возраста, гипогликемия). Изменения внешности наиболее выражены в младенчестве и в детстве, по мере взросления они становятся менее заметными. Наиболее часто сопутствующими новообразованиями являются опухоль Вильмса, гепатобластома, рабдомиосаркома, адренокортикальная карционома и нейробластома.
В некоторых случаях диагноз ставится лишь во взрослом возрасте, т.к. экспрессия генов крайне вариабельна. Даже у пациентов без выраженных визуальных признаков повышен риск развития онкологических заболеваний. При наличии у ребенка характерного фенотипа следует быть крайне настороженным в отношении возможного опухолевого роста и вовремя проводить необходимые диагностические процедуры: детальный осмотр и УЗИ внутренних органов каждые 6-12 недель до восьми лет; измерение уровней альфа-фетопротеина каждые 6-12 недель до пяти лет. Из всех новообразований на голове и шее чаще всего локализуется рабдомиосаркома, но, к счастью, даже в этой группе пациентов рабдомиосаркома встречается редко.
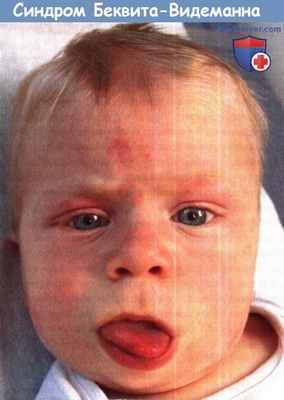
Макроглоссия и макросомия являются
характерными признаками синдрома Беквита-Видеманна.
Неотложная медицинская помощь требуется в нескольких возможных ситуациях. Дефекты передней брюшной стенки встречаются в 60-75% случаев, выраженные аномалии подлежат коррекции вскоре после рождения. Макроглоссия наблюдается в 80% случаев, иногда необходима трахеотомия. Макроглоссия обусловлена гипертрофией/гиперплазией как тела, так и корня языка, из-за чего уменьшение только передней его части не всегда достаточно для улучшения дыхания.
Если симптомы обструкции дыхательных путей при макроглоссии выражены умеренно, возможно динамическое наблюдение за пациентом без агрессивного хирургического лечения, т.к. иногда симптомы улучшаются самостоятельно вследствие опережающего роста костей лицевого скелета. У некоторых пациентов с макроглоссией и затруднением дыхания облегчения симптомов можно достигнуть проведением аденотонзиллэктомии.
Хирургическое лечение по поводу выраженного стеноза дыхательных путей необходимо проводить как можно раньше, т.к. оно помогает снизить риск таких осложнений, как развитие легочного сердца, нарушений сосания, и даже преждевременной смерти.
Макроглоссия часто является причиной дополнительных сложностей в формировании речи и правильной артикуляции. Дети с синдромом Беквита-Видеманна обычно имеют нормальный коэффициент умственного развития, но одной речевой терапии зачастую недостаточно для формирования языковых навыков; иногда требуется прибегать к хирургической коррекции языка.
Из-за выступающего вперед языка дети могут подвергаться оскорблениям со стороны сверстников, для достижения наилучшего косметического результата рекомендуется оперативное лечение. Проблемы с питанием, которые могут возникнуть из-за макроглоссии, возможно решить консервативным путем: приемом жидкой пищи, использованием бутылочек с крупным соском.
При выраженной асимметрии лица иногда требуется хирургическая коррекция, но в большинстве случаев визуальный дефект выражен весьма незначительно и становится незаметным по мере взросления.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Беквита-Видемана
Синдром Беквита-Видемана — это сложная мультигенная патология, которая проявляется множественными врожденными пороками развития. Заболевание возникает при различных генетических аномалиях 11 хромосомы в сочетании с эпигенетическими нарушениями. Триада признаков болезни Беквита-Видемана включает макроглоссию, омфалоцеле, макросомию. Также наблюдаются разнообразные соматические патологии. Диагностика представлена генетическим исследованием, визуализацией внутренних органов методами УЗИ, МРТ, КТ. План лечения и реабилитации подбирается индивидуально, в соответствии с тяжестью синдрома, числом и характером клинических симптомов.
МКБ-10
Q87.3 Синдромы врожденных аномалий, проявляющихся избыточным ростом [гигантизмом] на ранних этапах развития
Общие сведения
Причины
Патология возникает, когда сочетаются генетические причины (различные мутации ДНК) и эпигенетические факторы (метилирование ДНК, биохимические изменения гистонов). До 85% случаев синдрома проявляются спорадически, а оставшиеся 15% наследуются от родителей по аутосомно-доминантному типу с неполной пенетрантностью. Нарушения структуры 11-й хромосомы у страдающих болезнью Беквита-Видемана подразделяются на следующие группы:
- Нарушение метилирования IC2. Самый частый вид эпигенетической мутации, который выявляется у 50% пациентов. В большинстве случаев такие расстройства появляются у детей без отягощенной наследственности.
- Отцовская однородительская дисомия. При этом варианте синдрома ребенок наследует обе хромосомы из конкретной пары от одного родителя, что чревато развитием тяжелых врожденных пороков.
- Мутация CDKN1C. Такая генная аномалия имеет 50% вероятность передачи от больной матери ребенку, спорадические мутации встречаются крайне редко.
- Гиперметилирование IC1 (H19). Этот тип эпигенетических нарушений составляет до 5% всех случаев синдрома Беквита-Видемана. Он бывает без делеции (спорадические варианты) или с делецией (наследуется от матери).
- Аномалии локуса 11p15. Менее 1% пациентов составляют люди с цитогенетически видимой дупликацией хромосомы или ее транслокацией/инверсией. Такие мутации в основном наследуются от отца и матери.
Патогенез
В основе всех возникающих дефектов лежат молекулярные нарушения в генах короткого плеча 11 хромосомы. Здесь располагаются центры импринтинга, которые отвечают за правильную экспрессию генов, полученных от матери с отцом. У здоровых людей эти гены контролируют рост органов и тканей, угнетают избыточную пролиферацию клеток. Соответственно при их мутации у плода происходят множественные органные нарушения, формируются врожденные пороки скелета.
Симптомы
Болезнь проявляется патогномоничной триадой симптоматики («большие» критерии), которая включает омфалоцеле (пупочную грыжу), макроглоссию (слишком большой язык), макросомию (крупный плод). К типичным клиническим признакам синдрома Беквита-Видемана относят пропорциональное увеличение длины верхней и нижней конечности с одной стороны (гемигиперплазию), расхождение прямых мышц живота, дополнительные складки на мочке уха.
Среди нарушений внутренних органов превалируют медуллярная дисплазия почек, цитомегалия фетальной коры надпочечников, висцеромегалия — увеличение размеров печени, селезенки, поджелудочной железы. Реже встречаются пороки развития сердца, врожденная кардиомиопатия, кардиомегалия. После рождения у младенца обычно выявляется тяжелая неонатальная гипогликемия с характерным симптомокомплексом.
Осложнения
Наличие эпигенетических мутаций создает предпосылки для развития эмбриональных опухолей. Самые распространенные типы онкопатологии у больных с синдромом Беквита-Видемана — гепатобластома, нефробластома, рабдомиокарцинома, аденокарцинома. Из-за множественных врожденных аномалий, которые усугубляются полиорганными нарушениями, у пациентов сохраняется высокая вероятность летального исхода. В детском возрасте умирает до 20% больных.
Диагностика
Чтобы установить клинический диагноз синдрома Беквита-Видемана, у пациента необходимо выявить как минимум 3 «больших» критерия либо 2 «больших» + 1 «малый». При сочетании магроглоссии и пупочной грыжи стоит рассматривать возможность заболевания, даже если другая типичная симптоматика отсутствует. Расширенный план диагностических мероприятий включает следующие методы:
- Молекулярно-генетическое тестирование. При проведении генетической диагностики учитывается возможность гетерогенности синдрома, поэтому исследования делают сразу на несколько типичных мутаций, эпигенетических аномалий. Все дети с проявлениями синдрома Беквита-Видемана также проходят обязательное кариотипирование.
- Инструментальная визуализация. Специфическим признаком синдрома считается висцеромегалия, для обнаружения которой выполняется УЗИ, КТ или МРТ органов брюшной полости. Для диагностики кардиомегалии показана эхокардиография, а сопутствующие нарушения работы сердца регистрируются на ЭКГ.
- Неврологический осмотр. Обследование новорожденного у детского невролога необходимо для исключения сопутствующих поражений ЦНС, родовых травм на фоне макросомии. В будущем требуется оценка когнитивных способностей, которые могли пострадать вследствие неонатальной гипогликемии.
Лечение синдрома Беквита-Видемана
Специфическая терапия болезни еще не разработана. Лечение проводится мультидисциплинарной командой врачей и направлено на устранение или уменьшение отдельных проявлений синдрома Беквита-Видемана. Согласно клиническим протоколам, в план оказания медицинской помощи при этой генетической патологии включаются:
- Коррекция гипогликемии. Это самый важный момент лечения в неонатальном периоде, поскольку нормальный уровень гликемии необходим для функционирования головного мозга. Коррекция выполняется 10% растворами глюкозы, при необходимости вводятся глюкокортикоиды.
- Химиолучевая терапия. Такое лечение назначается при онкологических заболеваниях, которые зачастую возникают у детей раннего возраста в результате наследственной предрасположенности.
- Пластическая коррекция. Участие ортопедов и пластических хирургов требуется для устранения деформаций и аномальной длины конечностей, восстановления правильных пропорций тела, повышения функциональных возможностей опорно-двигательного аппарата.
С учетом типичных признаков и возможных осложнений синдрома разрабатывается комплексная программа диспансерного наблюдения пациента, для чего привлекают ортопедов, эндокринологов, неврологов, кардиологов. По показаниям применяются реабилитационные мероприятия с участием дефектологов, логопедов, специалистов ЛФК и массажистов.
Прогноз и профилактика
Синдром Беквита-Видемана сопряжен с высоким риском инвалидизирующих осложнений, а у 1/5 пациентов наблюдается летальный исход. Прогноз сомнительный, он определяется степенью выраженности фенотипических изменений, числом врожденных соматических патологий. Поскольку болезнь принадлежит к категории мультигенных с преимущественно спорадическим развитием, эффективные меры профилактики пока не разработаны.
2. Тактика амбулаторного наблюдения за пациентом с синдромом Беквита-Видемана/ Э.А. Каширина, А.А. Рубцова, Н.М. Югай, О.Б. Карабанова// Медицинский совет. — 2017. — №19.
3. Макросомия плода: современное состояние проблемы/ Р.С. Геворкян, А.Н. Рымашевский, А.Е. Волков, В.В. Маркина// Современные проблемы науки и образования. — 2016. — №6.
4. Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай)/ Т.А. Назаренко, Н.А. Зыряева// Проблемы репродукции. — 2014. — №3.
Макроглоссия (Глоссоцеле, Лопатообразный язык, Мегалоглоссия)
Макроглоссия — это врожденное или приобретенное увеличение языка. Гипертрофия может быть диффузной или частичной. Язык массивный, толстый, часто выдается наружу. При тяжелой макроглоссии возникают эпизоды апноэ во сне, нарушаются функции глотания и речи. Обследование включает в себя осмотр с определением размеров нижней челюсти, вспомогательное УЗИ и рентгенологические исследования. При симптоматической форме проводится медикаментозное лечение основного заболевания. Для уменьшения языка показано его клиновидное иссечение. При макроглоссии дезонтогенного происхождения проводят склеротерапию.
Общие сведения
Макроглоссия (глоссоцеле, широкий язык, лопатчатка, мегалоглоссия) — это патологическое увеличение или утолщение всего языка или отдельных его частей (кончика, спинки, боковых поверхностей). Из всех заболеваний языка макроглоссия выявляется в 15% случаев. При патологиях эндокринной системы распространенность гипертрофии достигает 33%. Выявлена отрицательная корреляция между глоссоцеле и ЛОР патологией. Половозрастных, географических особенностей заболеваемости не зафиксировано.
Причины макроглоссии
Врожденные дефекты
Глоссоцеле может быть врожденным или приобретенным. Основные причины истинной врожденной макроглоссии — наследственные заболевания, опухоли дезонтогенной природы. Идиопатическая мышечная мегалоглоссия неустановленной этиологии у плода встречается редко. Причинами врожденной макроглоссии могут быть:
- Генетические синдромы. Гипертрофия языка — один из характерных признаков синдромов Дауна, Симпсона-Голаби-Бемеля, Беквита-Видемана. Обнаружение мегалоксии у плода может указывать на мукополисахаридоз. При синдроме Помпе глоссоцеле встречается в 65% случаев.
- Врожденные опухоли. Макроглоссия у плода может развиться при опухоли дезонтогенного генеза — лимфангиоме. Гипертрофия языка возникает из-за повышенного разрастания лимфатических сосудов.
- Врожденный гипотиреоз. Отсутствие или недоразвитие щитовидной железы плода приводит к врожденному гипотиреозу с дефицитом или аномальной выработкой гормонов щитовидной железы. У младенцев с гипотиреозом диагностируется широкий увеличенный язык.
Факторы риска образования макроглоссии плода:
- наличие в семье случаев рождения ребенка с генетической патологией;
- возраст матери старше 35 лет;
- дефицит йода при беременности;
- токсическое действие свинца на плод.
Приобретенные причины
Патология развивается после рождения ребенка, не связана с генетическими мутациями или агрессивным влиянием на плод внешних факторов при внутриутробном развитии. К основным причинам приобретенной макроглоссии можно отнести:
- Травмы. Повреждение языка — причина временной макроглоссии. Увеличение органа происходит из-за кровоизлияния в мышечную ткань, посттравматического отека.
- Эндокринопатия. При мегалоксии возникают акромегалия и микседема. На фоне гиперпродукции гормона роста при акромегалии наблюдается гипертрофия мышечной ткани. При микседеме развивается отечная мегалоксия.
- Амилоидоз. Для него характерно поражение мышц языка из-за отложения патологических белков в тканях. Выраженные гипертрофические изменения диагностируются у каждого пятого пациента.
- Хронические инфекционные заболевания. В эту группу входят туберкулез и третичный сифилис. Глоссоцеле также может сигнализировать о грибковых заболеваниях — актиномикозе и бластомикозе.
- Новообразования. Развитие макроглоссии связано с инфильтрацией мышечной ткани раковыми клетками. При осмотре выявляется очаговая мегалоксия с локализацией, соответствующей пораженному участку.
- Ангионевротический отек. Причина макроглоссии — острая аллергическая реакция. После купирования приступа аллергии отек языка спадает.
Патогенез
Из-за генетических мутаций при наследственных заболеваниях Беквита-Видемана, Симпсона-Голаби-Бемеля увеличивается пролиферация клеток, катализируются факторы роста. Это приводит к развитию у плода гигантизма и макроглоссии. При мукополисахаридозе из-за дефицита лизосомальных ферментов катаболизм гликозаминогликанов нарушается. Отложение последнего в мышечной ткани плода является причиной макроглоссии.
Мегалоксия при лимфангиоме считается пороком развития лимфатических сосудов плода во время эмбриогенеза. При гипотиреозе чрезмерное накопление глюкозаминогликанов приводит к муцинозному отеку, отеку дермы и подкожно-жировой клетчатки.
Акромегалия возникает при гиперпродукции гормонов гипофиза. Наличие новообразования — аденомы гипофиза — увеличивает уровень гормона роста, который отвечает за рост и увеличение мягких тканей. Изменения амилоидоза вызваны внеклеточным отложением нерастворимых патологических фибриллярных белков.
Классификация
По времени и причинам развития различают врожденную и приобретенную макроглоссию. Даже в стоматологии истинная и ложная мегалоксия (родственник.
- Истинная форма характеризуется объективным увеличением размера языка.
- Ложное глоссоцеле выявляется при уменьшении ротовой полости, отсутствии резцов нижней челюсти или при инфильтрации раковых клеток дна ротовой полости.
В зависимости от степени вовлечения мышечной ткани языка в патологический процесс различают 2 формы:
- Диффузный — характеризуется равномерной гипертрофией всего органа. Чаще всего возникает у детей с генетическими заболеваниями, с нарушениями эндокринной системы, из-за острой аллергической реакции.
- Частичный — сопровождается очаговым утолщением кончика, корня или спинки, а также асимметрией языка. Причинами частичной макроглоссии являются травмы, специфические инфекционные заболевания, опухоли.
Симптомы макроглоссии
Врожденная макроглоссия
У новорожденного с глоссоцеле массивный язык не помещается в ротовую полость, постоянно торчит, рот приоткрыт. По бокам в области коренных зубов видны следы зубов, отмечается слюноотделение. Гипертрофия органов широко распространена. У младенцев мегалоглоссия затрудняет грудное вскармливание. В дальнейшем возникают проблемы с жеванием, суставом, развитие зубочелюстных деформаций. Другие сопутствующие симптомы зависят от основной патологии.
При генетически обусловленных заболеваниях Симпсона-Голаби-Бемеля и Беквит-Видеманна у детей возникают трудности с глотанием и дыханием. При прорезывании зубов образуется мезиальный прикус. Синдром Дауна характеризуется увеличенным складчатым «географическим языком».
При диффузной лимфангиоме язык увеличен, имеет луковичное утолщение переднего отдела и отпечатки зубов. На слизистой оболочке видно больше пузырьков. Рот больного ребенка приоткрыт. Открытый прикус образуется при чрезмерном развитии нижней челюсти и подбородка.
Приобретенная макроглоссия
Приобретенная форма макроглоссии развивается при жизни на фоне другого заболевания или травмы. У пациентов с акромегалией нижняя челюсть выдвигается вперед, из-за большого размера язык высовывается из полости рта. На спине наблюдается гипертрофия сосочков; также могут быть бороздки, складки.
Бластомикоз возникает с образованием узлов в толще слизистой оболочки. Из-за воспалительного инфильтрата язык при актиномикозе выглядит голубоватым и «деревянистым». Третичный сифилис характеризуется увеличенным твердым языком с дополнительными рубцами.
Макроглоссия при туберкулезе развивается медленно. Гипертрофии предшествует образование туберкулезного узла в толще мышечной ткани. Воспалительной реакции нет. Пациенты не жалуются на боли. При амилоидной макроглоссии язык плотный, сосочки гладкие, текстура восковая. Размер можно увеличить вдвое. На боковых поверхностях снимают оттиски зубов.
Осложнения
Новорожденный с мегалоссами может страдать от апноэ во сне, состояния, сопровождающегося кратковременной остановкой дыхания. Сустав поражен, часто диагностируется дисфагия — нарушение глотания. Чрезмерное давление языка на передние зубы — причина образования у ребенка патологического прикуса.
Клиническое обследование показывает прогрессирование (массивная нижняя челюсть выступает вперед) без окклюзионного контакта между передними, а иногда и боковыми зубами верхней и нижней челюсти. Поскольку у пациентов постоянно открыт рот, наблюдается сухость слизистой оболочки губ с последующим образованием трещин. Присоединение вторичной инфекции приводит к развитию воспаления.
Диагностика
Поскольку макроглоссия в подавляющем большинстве случаев носит симптоматический характер, требуется комплексное обследование. Во время клинического осмотра оценивается форма и размер языка. Чтобы отличить истинную патологию от ложной, определяется размер нижней челюсти. Ребенок осмотрен педиатром, челюстно-лицевым хирургом, генетиком. Дополнительные методы:
- Ультразвуковая диагностика. УЗИ при беременности позволяет обнаружить макроглоссию у плода даже в матке. После рождения для исключения гипотиреоза малышу назначают УЗИ щитовидной железы. При наличии пальпируемых образований в толще органа проводится УЗИ языка.
- Рентгенологическая диагностика Для подтверждения наследственной патологии используется нормальная рентгенография лицевого и мозгового черепа. Эзофагография — контрастная рентгенография пищевода — проводится для анализа функции глотания.
- МРТ и КТ полости рта. МРТ делают при подозрении на опухолевый процесс. КТ языка назначают в неотложных ситуациях детям раннего возраста при тяжелой дисфункции глотания, дыхательной недостаточности.
- Лабораторное исследование. Анализ крови на уровень гормонов помогает исключить эндокринную патологию у ребенка. Неонатальный скрининг на врожденный гипотиреоз позволяет выявить аномалии у новорожденных. Специфические лабораторные исследования предназначены для диагностики хронических инфекционных заболеваний (туберкулез, сифилис).
Лечение макроглоссии
Консервативная терапия
Консервативное лечение эффективно при выявлении преходящей макроглоссии у ребенка. Если мегаглоссия развилась из-за эндокринной дисфункции или гиперфункции, специфических воспалительных заболеваний (сифилис, туберкулез), то показано медикаментозное лечение основной патологии. Больным глосситом из-за присоединения вторичной инфекции назначают антисептические полоскания, местные противовоспалительные препараты, антибиотики. При обнаружении кандидозной инфекции используются противогрибковые препараты.
Хирургическое лечение
Наличие у ребенка апноэ во сне, нарушений глотания, суставов — прямые показания к хирургическому вмешательству. При истинной гипертрофии выполняется клиновидное иссечение части языка. У пациентов с изолированной формой патологии неясной этиологии лигируют язычную артерию. Ограничение кровоснабжения частично приостанавливает гипертрофию органов, но использование этого метода вызывает споры.
При диффузной опухоли — лимфангиоме — склеротерапию применяют с 2-3-кратным введением 2 мл 70% этилового спирта. Иногда назначают короткофокусную лучевую терапию.
В послеоперационном периоде контролируют степень отека, контролируют дыхание с оценкой сердечной деятельности. Во избежание инфицирования раневой поверхности назначают антибиотики. Во время реабилитации с ребенком работает логопед. Также показаны консультации специалиста по сну, ортодонта.
Прогноз и профилактика
Прогноз зависит от этиологии. Правильное лечение сопутствующей патологии в сочетании с хирургической пластикой дает удовлетворительные результаты. Уже в течение первых 3-4 месяцев после операции восстанавливается внешнее дыхание и глотание, улучшается разборчивость речи. Специфических мер первичной профилактики макроглоссии не разработано. Вторичная профилактика основана на соблюдении элементарных правил гигиены полости рта.
Читайте также:
- Острые и хронические фотодерматозы
- Весенний насморк или об аллергическом рините
- Целомические перикардиальные кисты. Распространенность перикардиальных кист
- Хорионический гонадотропин (ХГЧ) для индукции овуляции. Беременность
- Позадипузырное клетчаточное пространство. Топография позадипузырного пространства.