Спинальные мышечные атрофии
Добавил пользователь Валентин П. Обновлено: 27.01.2026
cпинальные мышечные атрофии (СМА) — генетически гетерогенная группа наследственных заболеваний центральной нервной системы (ЦНС), характеризующихся дегенерацией и гибелью двигательных нейронов передних рогов спинного мозга. Болезнь в основном выражается в постепенном развитии симметричных вялых параличей и атрофии поперечнополосатой мускулатуры, качественном перерождении соответствующих мышц и снижением их электровозбудимости [1].
Общепопуляционная распространенность СМА приблизительно равна 8,5—10,3 на 100 000 живых новорожденных, распространенность носительства составляет от 1 на 60 до 1 на 35 случаев. Так как наиболее часто СМА вызываются мутациями, носящими аутосомный характер наследования, соотношение пациентов обоих полов примерно равное.
СМА в Самарской области
Диагноз G12.1, кодирующий заболевания из группы «Другие наследственные спинальные мышечные атрофии», включающий прогрессирующий бульбарный паралич Фацио—Лонде, СМА взрослых, детскую СМА II типа, дистальные СМА, СМА III типа, Кугельберга—Веландер и скапулоперонеальную форму, установлен у 37 пациентов, в том числе у 12 детей. Из них 16 пациентов женского пола и 21 — мужского; младший пациент 2014 г. рождения, старший — 1945 г.
Диагноз G12.8 «Другие спинальные мышечные атрофии и родственные синдромы» установлен у 13 пациентов, в том числе — 1 ребенка 2007 г. рождения; основная часть этих пациентов рождены после 1970 г. Из них 5 женщин и 8 мужчин.
Диагноз G12.9 «Спинальная мышечная атрофия неуточненная» установлен у 2 взрослых пациентов — мужчины и женщины 1968 и 1986 г. рождения, проживающих в Самарской области.
Наряду со спинальными амиотрофиями группа G12 включает в себя код G12.2, которым обозначают боковой амиотрофический склероз (БАС). Часть проявлений этого заболевания схожа с таковыми у пациентов со СМА. БАС встречается чаще — в Самарской областной клинической больнице им. В.Д. Середавина по состоянию на июнь 2019 г. наблюдали 142 пациента.
Этиология и патогенез СМА
СМА возникают из-за недостатка белка выживаемости мотонейронов (SMN, БВМН) и других полипептидов, взаимодействующих с ним в норме, которые кодируются несколькими десятками генов. Большая часть СМА, как правило, вызывается мутациями в гене SMN1, находящемся на длинном плече 5-й хромосомы в 3-м бэнде 1-й области. Ген SMN1 продублирован располагающимся чуть ближе к области центромеры геном SMN2. Каждый из этих генов имеет несколько копий: SMN1, как правило, 2 — по одной на каждую хромосому из пары у представителей всех рас, за исключением афроамериканцев, у которых их более 2 [9].
Между количеством копий генов SMN1 и SMN2 и течением заболеваний группы СМА имеется корреляция — чем больше количество SMN2, тем позже манифестирует заболевание и легче его течение: при всех поврежденных копиях гена SMN1 у детей со СМА I типа в среднем от 1 до 2 неповрежденных копий гена SMN2, у пациентов со СМА II и III типов по крайней мере, 3 копии, у пациентов со СМА IV типа как минимум 4 копии гена SMN2. Отсутствие у зародыша сохранных копий SMN1 и SMN2 приводит к спонтанному аборту. Течение заболеваний зависит не только от количества копий гена SMN2, но и от других факторов, о чем говорит отсутствие строгой корреляции [10, 11].
По структуре ген SMN2 отличается от SMN1 одним нуклеотидом в 7-м экзоне, являющимся энхансером сплайсинга, что приводит к утрате его функции [12]. SMN2 по вышеуказанным причинам без вмешательств извне не способен обеспечить продукцию достаточного количества БВМН, который выполняет ряд критически важных функций: на эмбриональной стадии развития он регулирует часть составляющих процессов нейрогенеза, в дальнейшем участвует в обеспечении активного транспорта, в частности транспорта мРНК в аксональных областях нейронов; играет одну из ключевых ролей в регуляции сплайсинга и транскрипции, а также связан с процессами функционирования цитоскелета и регенерации теломер [13—15].
БВМН встречается как в цитоплазме, так и в ядре, где выполняет свои функции при помощи БВМН-взаимодействующих белков (SMN-interacting proteins): регулятора апоптоза Bcl-2, коилина, АТФ-зависимой геликазы А, фибрилларина, «вверх по течению» элемент-связывающий белок 1 (FUBP1), H/ACA рибонуклеинопротеидного комплекса субъединица 1 (GAR1), белков семейства GEMIN, гетерогенного ядерного рибонуклеопротеина R (HNRNPR), импортирующей субъединицы бета-1 (KPNB1), белка p53 и малых ядерных рибонуклеопротеинов Sm D1 и D2 (SNRPD1 и SNRPD2) [16—27]. Дефицит БВМН и взаимодействующих с ним белков может проявляться гипорефлексией, снижением мышечного тонуса и массы, мышечной слабостью, синдромом «вялого ребенка», отставанием или регрессом в моторном развитии, слабым кашлем, слабыми криком и плачем, респираторным дистресс-синдромом, фасцикуляциями языка, бульбарным синдромом [28].
Есть случаи присутствия мутаций других генов, вызывающих дополнительные морфологические и клинические проявления. Мутации, приводящие к СМА, наследуются в основном аутосомно-рецессивно, но часть из них имеет аутосомно-доминантный и X-сцепленный тип наследования [29].
При наиболее часто встречающейся причине СМА, мутации гена SMN1, наличие даже большого количества копий гена SMN2 не гарантирует производства должного уровня БВМН, но именно наличие неповрежденных копий SMN2 в геноме пациента со СМА способствует его выживанию и дает возможность проведения специфической генной терапии.
Классификация и диагностика СМА
Нозологии, входящие в группу СМА, традиционно различаются по степени выраженности и степени прогрессирования дегенераций и клинических проявлений, а также по их топике; по характерности манифестации в определенном периоде; по сопутствующим специфическим и параспецифическим нарушениям. На основании этих признаков формируется приблизительная классификация СМА, которая включает СМА взрослых, к ним относят СМА IV типа (OMIM271150), бульбоспинальную амиотрофию Кеннеди (OMIM 313200), аутосомно-доминантную сегментарную СМА (OMIM 183020), аутосомно-доминантную скапулоперонеальную СМА Старка—Кайзера (OMIM 181400), СМА Джокела (OMIM 615048), аутосомно-доминантную позднюю СМА Финкела (182980). Манифестация СМА взрослого возраста наступает на третьем или последующих десятках жизни пациентов.
Детские СМА, манифестирующие приблизительно до 18 лет, включают заболевания проксимальной, дистальной и бульбоспинальной подгрупп. К проксимальной подгруппе относят СМА I—III типов (MIM 253300, MIM 253550, MIM 253400), СМА Рюкю (OMIM 271200), СМА с понтоцеребеллярной гипоплазией (OMIM 607596), СМА с прогрессирующей миоклонической эпилепсией (OMIM 159950). Согласно результатам работ американских исследователей [30, 31], из 100 живых детей с проксимальной СМА 58 имеют проксимальную СМА I типа, 29 — II типа, и 13 III типа.
К дистальной подгруппе относят СМА с преимущественным поражением нижних конечностей — СМА 1 и 2А (OMIM 158600, OMIM 615290), дистальную скапулоперонеальную СМА (OMIM 181405), аутосомно-рецессивную дистальную СМА с респираторным дистресс-синдромом (OMIM 604320). К бульбоспинальной подгруппе относится синдром Брауна—Виалетто—Ван Лэре (OMIM 211530, OMIM 614707) и синдром Фацио—Лонде (MIM 211500).
Часть заболеваний, относящихся к СМА, проявляются внутриутробно и приводят к смерти уже до или вскоре после рождения, их обозначают как врожденные летальные СМА. К ним относzтся Х-сцепленная летальная СМА (OMIM 616866) и проксимальная СМА I типа с врожденными переломами (OMIM 616867).
Для диагностики СМА используется комплекс методов обследования, включающий генеалогический анализ, неврологический осмотр, электронейромиографию (ЭНМГ) и молекулярно-генетические методы исследования с применением мультиплексной лигазной реакции c последующей амплификацией, которая позволяет точно выявить количество копий генов SMN1 и SMN2, что важно при уточнении генотипа каждого отдельного пациента или носителя [1]. При постановке диагнозов группы G12 в Самарской областной клинической больнице им. В.Д. Середавина использовали в основном данные, полученные в результате анализа жалоб, личного и семейного анамнеза пациентов, особенностей клинической картины и показаний, собранных при проведении магнитно-резонансной томографии и ЭНМГ с накожными и игольчатыми электродами. Для уточнения диагноза и оценки курабельности каждого случая всем пациентам было рекомендовано проведение генетической экспертизы. В последнее время приоритетным диагностическим методом при подозрении на СМА является генетическое тестирование.
Лечение СМА
После постановки диагноза определяют объем медицинской помощи и разрабатывают эффективный план лечения, подразумевающий симптоматическую коррекцию сопутствующих нарушений, которые могут выражаться дыхательной недостаточностью, гастроэзофагеальным рефлюксом, метаболическими нарушениями. Пациенты в среднем 1 раз в 3—6 мес проходят многопрофильный мониторинг при участии пульмонолога, гастроэнтеролога, диетолога, ортопеда, реабилитолога, медицинского генетика и педиатра.
Необходимый объем медицинской помощи определяется по функциональному состоянию пациента. Для его оценки используют разработанную европейскими специалистами по нервно-мышечным заболеваниям классификацию больных по функциональному статусу:
— дети, которые не могут сидеть без посторонней помощи («лежачие пациенты»);
— дети, которые могут самостоятельно сидеть, но не могут ходить без посторонней помощи («сидячие пациенты»);
— дети, которые могут самостоятельно ходить («ходячие пациенты») [32, 33].
Лежачим пациентам для исследования степени выраженности дыхательных нарушений проводят физикальный осмотр с оценкой акта дыхания и эффективности откашливания, кардиореспираторный мониторинг и полисомнографию для выявления признаков гиповентиляции в состояниях бодрствования и сна; пульсоксиметрию для контроля оксигенации крови; рентгенографию грудной клетки в динамике. Также проводится мониторинг частоты инфекционных заболеваний респираторного тракта и антибиотикотерапии за последние 6 мес. При остром необъяснимом ухудшении дыхательных функций и повторных пневмониях для поиска причин проводят изучение функции глотания.
К мероприятиям мониторинга у сидячих пациентов добавляются осмотр ортопеда и рентгенологическая оценка динамики деформаций костей туловища. Пациентам, способным самостоятельно ходить, также проводят исследование функции внешнего дыхания и регулярную спирометрию.
Оценка гастроэнтерологической патологии включает поиск ранних симптомов гастроэзофагеального рефлюкса, выполнение эзофагогастродуоденоскопии для оценки возможности установки зонда, обнаружения аномалий строения и подтверждения наличия рефлюкса. Исследование моторики, включая лучевую диагностику, позволяет подтвердить задержку эвакуации содержимого желудка, что может усугублять течение гастроэзофагеального рефлюкса. Метаболические и ортопедические нарушения менее опасны для жизни пациентов, их мониторинг основан на оценке антропометрических показателей и функциональных физических пробах.
По результатам мониторинга устанавливается объем необходимой помощи в виде частоты и кратности применения очистки дыхательных путей аспиратором, неинвазивной вентиляции легких с постоянным положительным давлением, постурального и прочих методов дренажа, кислородотерапии, применения антибактериальных средств, муколитиков и бронходилататоров или, при неэффективности, интубации и искусственной вентиляции легких. Также решается вопрос о целесообразности применения антацидов, блокаторов протонной помпы и прокинетиков при гастроэзофагеальном рефлюксе и нарушениях моторики желудочно-кишечного тракта; утверждаются объемы использования ноотропных, нейро- и кардиопротекторных, метаболических и прочих средств; ортопедической, в том числе хирургической, коррекции соответствующих патологий [34].
Существование таких пациентов в семье требует постоянного специфического ухода, связанного в первую очередь с предотвращением нарушений со стороны дыхательной системы, для чего существуют специально разработанные методики укладывания детей со СМА. Большинству пациентов необходима помощь в откашливании и удалении легочного секрета, для этого ухаживающие могут использовать постуральные и мануальные техники, вроде дренирующего массажа, при необходимости они могут воспользоваться механическим откашливателем или аспиратором. Использование аппарата неинвазивной вентиляции легких с постоянным положительным давлением в домашних условиях также контролируется ухаживающим лицом. Эффективность ухода может оцениваться по показателям пульсоксиметрии и длительности отсутствия осложнений со стороны дыхательной системы. Уход за пациентами со СМА также включает в себя следование программе диетотерапии и связанным с кормлениями рекомендациям по размещению пациента, использование максимума функциональных возможностей ортопедических средств для удобства и безопасности пациента, а также выполнение прочих назначений [35].
Перспективы симптоматической коррекции несравнимы с таковыми у препаратов патогенетической терапии. Сегодня существует 2 одобренных FDA патогенетических препарата для СМА, вызванной повреждением гена выживаемости мотонейронов-1 (SMN1), еще один проходит третью фазу клинических испытаний, исследования еще 2 находятся во второй; разрабатываются и готовятся к испытаниям еще несколько препаратов.
Второй препарат, одобренный FDA в мае 2019 г., AVXS-101, разработан компанией AveXis, принадлежащей Novartis. Он представляет собой раствор, содержащий вирусные векторы, заполненные геномом вируса, за исключением участка ДНК, необходимого для самовоспроизводства, и нативными копиями гена SMN1 с промоторами [37]. Пациентам препарат вводят внутривенно. Вопрос кратности введения обсуждается. На настоящий момент известно, что после единичной внутривенной инъекции AVXS-101 клиническое ухудшение не наступает как минимум 2 года; есть основания полагать, что длительность терапевтической активности препарата может оказаться гораздо дольше [38]. Цена дозы препарата на момент регистрации составляла более $2 млн и в настоящее время практически не снизилась [39].
Существует класс экспериментальных патогенетических препаратов, которые входят в класс «малых молекул», представляющих собой производные кумаринов, изо-кумарина и пиридо-пиримидинонов [40]. Перспективность использования обусловлена способностью «малых молекул» селективно модифицировать сплайсинг SMN2 при пероральном приеме. К этой группе патогенетических препаратов относится Рисдиплам, разработанный компанией F. Hoffmann-La Roche в сотрудничестве с PTC Therapeutics и НКО SMA Foundation и сейчас проходящий третью фазу клинических испытаний [41].
На сегодняшний день в России возможно использование потенциально доступных медико-генетической службе РФ методов, способных в ближайшем будущем снизить число пациентов со СМА и другими наследственными заболеваниями, что имело бы много положительных последствий, в том числе связанных со снижением нагрузки на бюджет МЗ РФ и уменьшением числа пациентов, попадающих в льготные социальные группы и получающих препараты бесплатно. Профилактическое использование молекулярно-генетической диагностики для пренатального выявления подобных заболеваний и их носительства у способных к деторождению взрослых в совокупности с бурно развивающимися технологиями генетического редактирования позволяет надеяться на крайне благоприятные прогнозы касательно будущего организации медицинской помощи с наследственными нервно-мышечными заболеваниями.
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
Сведения об авторах
Как цитировать:
Врач СПбГПМУ рассказала об опыте лечения спинальной мышечной атрофии

Спинальная мышечная атрофия (СМА) – генетическая болезнь, которая проявляется как нарастающая мышечная слабость. У пациента прогрессивно снижается способность мышц к сокращению, что проводит в итоге к нарушению дыхания и глотания. При этом интеллект абсолютно сохранен, и человек осознаёт все происходящие с ним страшные изменения.
Каждый год в России рождается около 200 детей с СМА. Существуют различные формы этого заболевания, наиболее тяжёлая – болезнь Верднига-Гоффмана – первые симптомы которой появляются в возрасте до 6 месяцев и быстро прогрессируют. Без респираторной поддержки такие малыши раньше редко доживали до двух лет, а врачи могли предложить им только паллиативную помощь.
Сегодня ситуация изменилась. Есть препараты, действие которых направлено на увеличение уровня функционального белка SMN – у людей, страдающих спинальной мышечной атрофией, он снижен. Этот белок необходим для работы двигательных нейронов спинного мозга. Если вовремя начать лечение, можно остановить гибель мотонейронов, а значит, избежать тяжёлых последствий заболевания.
Об опыте применения первого перорального препарата для лечения СМА в Педиатрическом университете рассказала невролог Диана Баюнчикова.
Малыши вместе с мамами находились в отделении патологии новорожденных и детей грудного возраста СПбГПМУ около 10 месяцев. В конце декабря одного ребёнка выписали домой, а второго перевели в стационар по месту жительства.
По словам врачей, сейчас состояние пациентов заметно улучшилось. На момент поступления в клинику Педиатрического университета, они могли двигать только кистями рук, нуждались в неинвазивной вентиляции лёгких и получали энтеральное питание. В настоящее время одна пациентка уже дышит самостоятельно и может глотать. Кроме того, малышка двигает руками и ногами, научилась хватать игрушки и активно тянуть их в рот. Серьезный прогресс наблюдается и у второго пациента, состояние которого изначально было тяжелее. У него тоже идет видимый прогресс в развитии двигательных навыков, правда, ещё сохраняется потребность в респираторной поддержке.
– Пока эти дети будут принимать препарат, ухудшений не будет. Насколько они реабилитируются, смогут ли вставать и самостоятельно ходить, сказать сложно. Но, по данным исследований, около 30% детей, которые получали препарат в течение года, смогли удерживать позу сидя, 83% – могли переворачиваться с боку на спину и смогли питаться самостоятельно. По опыту зарубежных коллег, пациентка, которая получает препарат уже 2,5 года и она научилась сама ходить с опорой к 3 годам , – рассказала Диана Баюнчикова.
Врач также сделала акцент на том, что помимо лекарственной терапии, огромную роль играет своевременная и полная реабилитация и ежедневные гигиенические процедуры дома.
- После приема препарата состояние ребёнка не изменится в один день. Это только начало пути, который предстоит родителям. Таким детям необходимо много медицинской аппаратуры после выписки. Обязательно наличие дома аспиратора, откашливателя, аппарата ИВЛ, инфузомата, мешка Амбу, и разных расходников. Очень здорово, что есть фонды, которые помогают таким семьям, нам очень сильно помогал фонд «Семьи СМА». Помимо материальной, фонд оказывает колоссальную информационную помощь родителям. Разумеется, детям необходим тщательный уход и различные процедуры – в том числе массажи, вибрационные массажи, двигательная реабилитация, позиционная терапия и другие мероприятия , - подчеркнула она.
Сакинат , мама одной из пациенток, рассказала, что возвращение в родной Надым после длительного лечения стало огромным счастьем и для неё, и для её младшей дочки Раяны. Три старших дочери Сакинат теперь буквально не отходят от сестры.
- С самого начала я не была уверенна, что все будет хорошо. Но надежда всегда есть, и положительный настрой был с самого начала. Надеялась, что все получится. Завышенную планку, что она пойдёт и будет развиваться как сверстники, я не ставлю. То, что ребёнок сам дышит, что за ней можно ухаживать дома при таком заболевании уже очень радует , - рассказала мама пациентки и добавила: «С того момента как мы начали получать препарат, болезнь перестала прогрессировать. С нашим диагнозом это огромный успех».

Как диагностируют спинально-мышечную атрофию?
При спинальной мышечной атрофии очень важно не опоздать с лечением. Именно бдительность мамы помогла остановить болезнь Раяны. Сакинат вовремя обратила внимание, что девочка, которая родилась абсолютно нормальной, стала апатичной и вялой.
- Она много спала, просыпалась только на кормление. Вроде бы для любой мамы удобно - ребенок спит и ест, спит и ест. Но, так как это у меня это уже четвёртый ребенок, я начала бить тревогу. И обратилась к участковому педиатру со своими опасениями , - рассказала женщина.
К счастью, врач внимательно отнёсся к жалобам мамы. После обследования в стационаре, ребёнка из Ямало-Ненецкого автономного округа отправили в Санкт-Петербург.
- Если вдруг у ребенка в раннем возрасте отмечается гипотония – то есть он малоподвижен, у него снижен тонус мышц, он мало двигается – то можно заподозрить нервно-мышечную болезнь. В этом случае необходима электронейромиография, а затем - генетическое исследование , - сообщила Диана Баюнчикова.
А вот в обязательный неонатальный скрининг диагностика спинально-мышечной атрофии пока не входит. Вопрос о необходимости внесения СМА в этот перечень пока только обсуждается.
Чем лечат спинальную мышечную атрофию?
Существует три препарата, которые применяются для лечения СМА.
В августе 2019 фармацевтическая компания Biogen зарегистрировала в России препарат «Спинраза» (нусинерсен). Препарат вводится в спинной мозг раз в несколько месяцев. Такие инъекции требуются пожизненно
В ноябре 2020 года регистрационное удостоверение получил «Эврисди» (рисдиплам) от компании Roche, который по принципу действия похож на Спинразу, но выпускается в виде раствора для приема в рот, поэтому его можно применять в домашних условиях.
В настоящее время также ведутся исследования, касающиеся комбинированной терапии, когда пациент начинает терапию одним из препаратов, а в дальнейшем ему вводится «Золгенсма».
– Все три препарата направлены на один результат. Они восстанавливают содержание функционального белка SMN в плазме крови человека. Соответственно, не происходит разрушение двигательных нейронов и мы не наблюдаем прогрессирование гипотонии. Это позволяет ребенку развиваться согласно возрасту. Но все это будет эффективно при условии, что терапия была инициирована в самом начале заболевания, пока не развились клинические признаки или они были незначительными. Чем позже, начнется лечение, тем сложнее восстанавливать двигательные функции, функцию дыхания и глотания. Также с течением заболевания появляются множественные полиорганные осложнения, которые часто носят не обратимый характер, - пояснила невролог СПбГПМУ.

В настоящее время 230 пациентов, которым необходим «Эврисди» получают его по программе раннего доступа к препарату. Со следующего года лечение детей со СМА будет финансироваться из фонда, созданного путем повышения НДФЛ до 15% для людей, чей доход превышает 5 млн рублей в год. Президент Владимир Путин уже подписал соответствующий закон.
Генетический тест на СМА
Спинальная мышечная атрофия (CMA) или проксимальная спинальная амиотрофия – это прогрессирующее наследственное заболевание, при котором происходит поражение клеток спинного мозга, приводящее к развитию слабости мышц, их атрофии, и в итоге, обездвиживанию пациента. Причиной заболевания в большинстве случаев являются мутации в гене SMN1, находящемся на длинном плече 5 хромосомы (5q).
Дети со спинальной мышечной атрофией рождаются с частотой 1 на 6000 рождений. Такой ребенок обычно рождается в семье, где оба родителя, и мать, и отец, как правило, здоровы, у них нет больных родственников, и никто в семье не ожидает рождение ребенка с унаследованным генетическим заболеванием. Это происходит, потому что тип наследования при СМА аутосомно- рецессивный и для развития болезни должны быть унаследованы 2 дефектных копии гена SMN1, от мамы и от папы. Сами родители при этом являются бессимптомными носителями мутации и до рождения больного ребенка, чаще всего, об этом не знают.
В России каждый 35-36-й человек является бессимптомным носителем мутации в гене SMN1.
В зависимости от возраста манифестации выделяют различные по тяжести формы CMA:
- І тип: Болезнь Верднига-Гоффмана,
- II тип: Промежуточная форма CMA,
- III тип: Болезнь Кугельберга-Веландер,
- IV тип: Взрослая форма CMA.
Диагноз спинальной мышечной атрофии может быть установлен на основании клинической картины заболевания в сочетании с данными генетического тестирования о наличии мутации в гене SMN1.
В настоящее время эффективных лекарственных средств, позволяющих вылечить детей со СМА нет, но есть препараты, которые способны значительно замедлить развитие болезни, продлить жизнь пациентам и улучшить ее качество.
Болезнь можно предупредить, если вовремя выявить генетический риск, то есть носительство мутации у здоровых родителей.
Диагностика скрытого носительства мутаций в гене SMN1 позволит при планировании беременности значительно снизить риск рождения в семье ребенка со спинальной мышечной атрофией.
Для семей, имеющих высокий риск рождения ребенка с наследственным заболеванием и желающих иметь здоровых детей, существует несколько вариантов профилактики:
- проведение инвазивной диагностики во время беременности,
- использование донорской спермы или яйцеклеток,
- преимплантационное генетическое тестирование (ПГТ).
ПГТ позволяет обследовать эмбрионы, полученные с помощью ЭКО, еще до беременности и выбрать тот, который не унаследовал СМА и другие заболевания. Все больше семей в мире предпочитают именно такой путь.
Узнать о риске, не дожидаясь рождения больного ребенка, возможно, пройдя в лаборатории Genetico исследование на носительство мутации в гене SMN1.
Всем людям, осознанно планирующим рождение ребенка.
Для диагностики при наличии симптоматики СМА.
При раннем выявлении заболевания у родственников.
Для проведения исследования необходимо сдать кровь из вены.
В день сдачи анализа не рекомендуется употреблять жирную пищу.
- Обращение в лабораторию Genetico.
- Забор биоматериала (кровь из вены).
- Исследование ДНК.
- Выдача лабораторного заключения.
Для проведения исследования необходимо сдать кровь из вены.
В день сдачи анализа не рекомендуется употреблять жирную пищу.
- Обращение в лабораторию Genetico.
- Забор биоматериала (кровь из вены).
- Исследование ДНК.
- Выдача лабораторного заключения.
В случае выявления гетерозиготного носительства генетический тест позволит избежать рождения больного ребенка со спинальной мышечной атрофией.
Спинальная амиотрофия типы I, II, . Поместите услугу в корзину сейчас, чтобы вернуться к ней позже В корзину
Медико-генетический центр и лаборатория
Широкий спектр услуг в области медицинской генетики

Спинальные мышечные атрофии (СМА)
Спинальные амиотрофии включают в себя несколько видов группы наследственных заболеваний, характеризующихся поражением скелетных мышц из-за прогрессирующей дегенерации нейронов передних рогов спинного мозга и двигательных ядер ствола головного мозга. Проявления могут начаться в младенчестве или детстве. Они варьируют в зависимости от типа заболевания и могут включать в себя гипотонию, гипофлексию, нарушение сосания, глотания и дыхания, задержку развития и в тяжелых случаях – раннюю смерть. Диагностика осуществляется при помощи генетического тестирования. Лечение носит поддерживающий характер.
Спинальные мышечные атрофии обычно являются результатом аутосомно-рецессивных мутаций, которые влияют на выживаемость мотонейрона 1 (SMN1) на длинном плече 5-й хромосомы, чаще всего вызывая гомозиготную делецию 7-го экзона. Спинальная мышечная атрофия может также поражать центральную нервную систему, поэтому спинальные амиотрофии не могут считаться заболеванием только периферической нервной системы. SMN2 является геном-модификатором; он на 99% идентичен гену SMN1 и расположен на длинном плече 5 хромосомы (5q); если SMN2 присутствует в нескольких копиях, то может изменить тяжесть заболевания и объяснить фенотипические различия у детей с СМА. Кроме того, существуют редкие формы СМА, которые не обусловлены 5q-мутациями.
Существует пять основных типов спинальной мышечной атрофии.
При спинальной мышечной атрофии типа 0 заболевание начинается в пренатальном периоде; оно проявляется в виде снижения подвижности плода на поздних сроках беременности и выраженной слабости и гипотонии при рождении. Больные новорожденные имеют лицевую диплегию, арефлексию, пороки сердца и иногда артрогрипоз. Смерть вследствие дыхательной недостаточности наступает в течение первых 6 месяцев.
Спинальная мышечная атрофия 1 типа (инфантильная спинальная мышечная атрофия, или болезнь Верднига-Гофмана) также присутствует внутриутробно и проявляется примерно в возрасте 6 месяцев. Отмечается мышечная гипотония (часто заметная при рождении), гипорефлексия, фасцикуляции языка и выраженные затруднения при сосании, глотании и дыхании. Смерть наступает от дыхательной недостаточности на первом году жизни в 95% случаев, и к 4 годам погибают все больные.
При спинальной мышечной атрофии 2 типа (промежуточная форма, или болезнь Дубовица), симптомы обычно проявляются в возрасте от 3 до 15 месяцев; 25% пострадавших детей учатся сидеть, но не ходят и не ползают. Развивается вялый паралич и фасцикуляции, что трудно выявить у маленьких детей. Выпадают глубокие сухожильные рефлексы. Может наблюдаться расстройство глотания. Большинство детей к 2-3 годам становятся прикованными к инвалидной коляске. Заболевание часто приводит к смерти в раннем возрасте от дыхательных осложнений. Однако прогрессирование заболевания может внезапно остановиться, но устойчивая слабость и высокий риск тяжелого сколиоза и его осложнений сохраняются.
Спинальная амиотрофия 3 типа (болезнь Кугельберга–Веландер) обычно проявляется в возрасте между 15 месяцев и 19 годами. Признаки похожи на симптомы I типа заболевания, но болезнь прогрессирует медленнее, а продолжительность жизни больше (иногда нормальна). Некоторые семейные случаи связаны с ферментными дефектами (например, дефицит гексозаминидазы). Симметричная слабость и атрофии, начинаясь с четырехглавой мышцы бедра и сгибателей бедра, постепенно распространяются дистальнее, становясь наиболее выраженными на голенях. Позже поражаются руки. Продолжительность жизни зависит от развития дыхательных осложнений.
Спинальная амиотрофия 4 типа (при позднем проявлении) может наследоваться по рецессивному, доминантному или Х-сцепленному типу; с дебютом в зрелом возрасте (30–60 лет) и медленно прогрессирующей слабостью и атрофией в основном проксимальных мышц. Это заболевание трудно отличить от амиотрофического бокового склероза Боковой амиотрофический склероз (БАС) и другие болезни мотонейрона (БМН) Боковой амиотрофический склероз и другие болезни мотонейрона характеризуются стойкой, непрерывной, прогрессирующей дегенерацией кортикоспинальных путей, клеток переднего рога, бульбарных двигательных. Прочитайте дополнительные сведения , поражающего главным образом нижние мотонейроны.
Спинальная мышечная атрофия (СМА): что это такое?
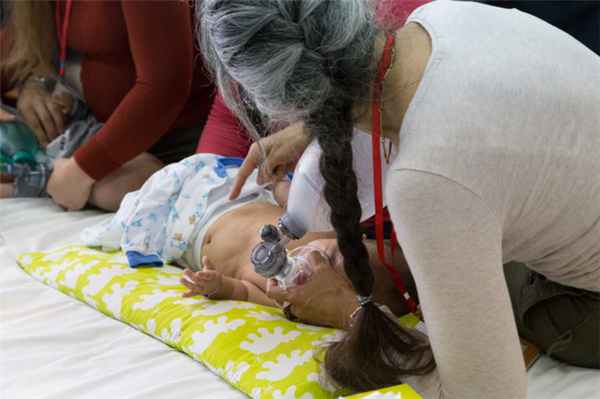
СМА — редкая генетическая болезнь, требующая участия специалистов паллиативной помощи. Какие степени СМА бывают, чем характеризуется каждая, какое оборудование нужно пациентам - читайте в статье
Спинальная мышечная атрофия (СМА): что это такое?
СМА — редкая генетическая болезнь, требующая участия специалистов паллиативной помощи. Какие степени СМА бывают, чем характеризуется каждая, какое оборудование нужно пациентам - читайте в статье
СМА – одно из самых часто встречающихся генетических заболеваний из разряда редких: оно встречается у одного новорожденного из 6-10 тысяч. Если в семье есть ребенок со СМА, он должен получать паллиативную помощь, поскольку заболевание прогрессирующее и на сегодня - неизлечимое. Однако степени заболевания очень разные: одни пациенты умирают во младенчестве, другие — живут до старости.
Предлагаем вам познакомиться с основной информацией о СМА и той помощи, в которой нуждается семья с этим редким заболеванием. Информацию собрал и упорядочил фонд "Семьи СМА": полную версию "ликбеза", включая истории семей, психологические аспекты, решение медицинских проблем в зависимости от состояния пациента, вы можете найти на сайте специального проекта фонда "Семьи СМА".
Что такое СМА?
СМА (спинальная мышечная атрофия) ― генетическое нервно-мышечное заболевание, которое поражает двигательные нейроны спинного мозга и приводит к нарастающей мышечной слабости. Заболевание носит прогрессирующий характер, слабость начинается с мышц ног и всего тела и с развитием заболевания доходит до мышц, отвечающих за глотание и дыхание. При этом интеллект больных СМА абсолютно сохранен.
В зависимости от тяжести симптомов выделяют 3 основных типа СМА: СМА 1, СМА 2, СМА 3. Чем раньше проявляются первые признаки болезни, тем ярче выражены симптомы, тем они тяжелее и тем быстрее прогрессирует заболевание.
СМА I (БОЛЕЗНЬ ВЕРДНИГА-ГОФФМАНА)
Наиболее тяжелая форма. Возраст проявления болезни: до 6 месяцев
Описание
- Выраженная мышечная гипотония; синдром «вялого ребенка»; не держит голову; не достигает способности сидеть и переворачиваться; обвисшее тело при удерживании подвешенным на животе;
- Ослабленные кашлевой, сосательный и глотательные рефлексы; поперхивание; дыхательные нарушения;
- В анамнезе может быть сниженная внутриутробная активность плода. Может наблюдаться деформация суставов и конечностей из-за внутриутробной гипотонии.
Течение
- Грубая задержка моторного развития;
- Быстрое развитие контрактур и деформаций грудной клетки;
- Прогрессирование бульбарных и дыхательных нарушений, проблемы с глотанием еды и слюны, отхождением мокроты;
- Высокий риск развития аспирационных пневмоний;
- Быстрое нарастание дыхательной недостаточности, особенно при присоединении инфекции.
Прогноз
- Наиболее тяжелая форма: при отсутствии респираторной поддержки большинство детей не доживают до 2 лет;
- Смерть наступает, как правило, из-за нарастания дыхательной недостаточности и развития пневмоний;
- Своевременная респираторная поддержка может увеличить продолжительность жизни ребенка;
- Такие дети нуждаются в паллиативном наблюдении.
«Нет человека без патологических генов». Генетик Наталия Белова рассказывает о редких болезнях и помощи семьям с больными детьми. А также о том, можно ли избежать генетической «поломки» у ребенка и почему жизнь «редких» — пока еще борьба
СМА II (БОЛЕЗНЬ ДУБОВИЦА)
Возраст проявления болезни: 6-18 месяцев.
Описание
- Отставание в моторном развитии;
- Способность сидеть без поддержки, иногда ― ползать или стоять, но эти возможности редуцируются по мере взросления;
- Может наблюдаться тремор пальцев;
- Мышечные и скелетные деформации;
- Нарушения дыхания.
Течение
- Задержка моторного развития, его остановка и регресс;
- Слабость межреберных мышц, поверхностное диафрагмальное дыхание, ослабление кашлевой функции, со временем развитие дыхательной недостаточности;
- Повышенный риск осложнений после респираторной инфекции;
- Деформации грудной клетки, контрактуры, сколиоз.
Прогноз
- Своевременная помощь и респираторная поддержка увеличивают продолжительность жизни.

Юлия Самойлова. Самый известный в России человек со СМА
СМА III (БОЛЕЗНЬ КУГЕЛЬБЕРГА-ВЕЛАНДЕРА)
Возраст проявления болезни: после 18 месяцев
Описание
- Способность ходить самостоятельно (со временем теряется);
- Сложности с комплексными моторными навыками (например, подъем по лестнице, бег);
- По мере прогрессирования болезни могут отмечаться трудности с жеванием и глотанием, а также дыхательные проблемы и проблемы с откашливанием.
Течение
- Прогрессирует медленно;
- К подростковому возрасту большинство больных садятся в коляску, но у некоторых способность самостоятельно ходить может сохраниться до взрослого возраста;
- Со временем появляются выраженные контрактуры и сколиоз;
- Риск осложнений после респираторной инфекции.
Прогноз
- При надлежащем уходе имеют обычную продолжительность жизни.
Механизм СМА
СМА вызывается поломкой в гене SMN1. Ген SMN2 частично компенсирует утрату гена SMN1.
- Ген SMN1 поврежден и не соответствует норме
- Важные белки не производятся в достаточном количестве
- Двигательные нейроны функционируют неправильно и отмирают
- Импульсы не распознаются
- Мышцы теряют силу и атрофируются
- Движения ограничены, это затрудняет перемещение, дыхание и глотание
Диагностика и помощь специалистов
При выявлении симптомов, которые могут указывать на болезнь, необходимо комплексное обследование для установления точного диагноза.
Диагностика: необходимо получить консультацию невролога, специалиста по нервно-мышечным заболеваниям, который может установить диагноз на основании симптомов. Для подтверждения диагноза требуется проведение ДНК-диагностики и консультация генетика.
Генетическая диагностика: тест ДНК для выявления делеции гена SMN1 и определения количества копий SMN2.
Дополнительные исследования:
- Биохимия крови: креатинкиназа (КФК) — в норме при СМА I, в норме или незначительно повышена при других типах;
- Электронейромиография — показывает снижение нервных импульсов, помогает дифференцировать СМА от других нервно-мышечных болезней. Сенсорная нервная проводимость обычно в норме.
Медицинская помощь
Врачи-специалисты:
- Невролог ― ставит диагноз, назначает поддерживающее лечение, ведет постоянное наблюдение за ходом болезни.
- Генетик ― ставит диагноз и, в случае необходимости, консультирует семью по вопросам дальнейшего потомства.
- Участковый педиатр (терапевт) ― помогает лечить болезни, которыми болеют все.
- Пульмонолог или реаниматолог ― помогает выявить и скомпенсировать дыхательные нарушения, решать проблемы с откашливанием, дает консультации по респираторной поддержке.
- Ортопед ― оценивает нарушения опорно-двигательного аппарата (контрактуры, деформации), определяет необходимый объем профилактических мер, помогает корректировать эти нарушения.
- Нейрохирург ― занимается исправлением сколиоза. ― подбирает комплекс упражнений и абилитационных процедур и обучает родителей регулярно делать их дома самостоятельно.
- Нутрициолог или диетолог ― помогает подобрать оптимальное питание.
- Гастроэнтеролог ― помогает в случае возникновения проблем с желудком и кишечником.
- Кардиолог ― наблюдает за работой сердечно-сосудистой системы.
- Специалист по паллиативной помощи ― помогает комплексно улучшить качество жизни.
- Другие специалисты привлекаются по мере возникновения специфических проблем.
Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчас Личная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
Важные аспекты:
- Семейно-ориентированный подход ― врач учитывает мнение семьи по всем вопросам, касающимся лечения ребенка, в том числе проведения медицинских вмешательств и их объема, их приемлемости и сроков проведения;
- Ориентация на качество жизни пациента ― прежде чем предложить семье применение каких-либо медицинских технологий, необходимо учесть, как это повлияет на качество жизни, потому что важно жить полноценно, а не существовать;
- Качественная коммуникация и полноценное информирование пациента и членов семьи обо всех аспектах СМА ― как можно более полная информация о болезни, о том, что будет происходить и с чем придется столкнуться, а также как с этим справляться;
- Обучение практическим навыкам ухода и применения медицинского оборудования должно быть неотъемлемой частью медицинской помощи;
- Междисциплинарный и мультипрофессиональный подход ― необходима работа междисциплинарной команды (к примеру, невозможно ограничиться наблюдением невролога и получить весь спектр необходимой помощи для полноценной поддержки).
Помогающие организации
Благотворительный фонд «Семьи СМА» — единственная в России организация, специализирующаяся на помощи семьям, столкнувшимся со спинальной мышечной атрофией. Оказывается благотворительную, информационную, психологическую поддержку семьям, консультирует специалистов по вопросам заболевания и методов работы с пациентами со СМА.
Фонд помощи хосписам «Вера». Благотворительная и консультативная помощь семьям с неизлечимо больными детьми, неизлечимо больным взрослым.
ОДКБ № 1 Отделение паллиативной помощи детям, Екатеринбург, Свердловская область. Оказывается медицинскую, информационную, социальную и психологическую помощь семьям, воспитывающим ребенка-инвалида с паллиативным состоянием.
"Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева" ФГБОУ ВО РНИМУ им.Н.И.Пирогова. Институт находится в Москве. Обращаться за медицинской помощью детям со СМА и другими нервно-мышечными заболеваниями могут жители всей России.
Клиника «Чайка». Консультации врача-пульмонолога Штабницкого Василия Андреевича для детей и взрослых со СМА.
Детский хоспис «Дом с маяком» (Москва, ближнее Подмосковье). Медицинская, психологическая, правовая, социальная, благотворительная помощь семьям с неизлечимо больными детьми и молодыми взрослыми (до 25 лет).
Марфо-Мариинский медицинский центр «Милосердие» (г.Москва). Медицинская, психологическая, правовая помощь, помощь няни, мероприятия, духовная поддержка, благотворительная помощь семьям с неизлечимо больными детьми.
Более подробную информацию вы найдете на сайте благотворительного фонда "Семьи СМА" и их специального проекта о жизни со спинальной мышечной атрофией. Спецпроект предназначен тем, кто столкнулся с диагнозом СМА и хотел бы узнать все самое важное об этой болезни: к каким специалистам и куда обращаться, как ухаживать, за чем следить, о чем помнить, какая терапия существует на сегодняшний день.
Читайте также: