Туберозный склероз у плода. Склероз Бурневилля
Добавил пользователь Morpheus Обновлено: 27.01.2026
Туберозный склероз у плода. Склероз Бурневилля
Данный синдром характеризуется ангиофибромой лица (часто неправильно называемой жировой аденомой), эпилепсией, задержкой умственного развития, кистами почек, множественными двухсторонними ангиофибромами почек и рабдомиомой сердца. Также типичным для данного синдрома является наличие различных кожных проявлений, таких как гипохромные пятна видимые под лампой Вуда (Wood) и пятна цвета «кофе с молоком» в виде светло-коричневых зон. Кроме этого, могут наблюдаться такие опухоли мозга, как эпендимома третьего желудочка и астроцитома.
Синонимы. Склероз Бурневилля (Bourneville). Распространенность. От 0,3 до 1,0 на 10 000 родов.
Этиология. Аутосомно-доминантный тип наследования с большой долей новых мутаций (спорадические случаи). Экспрессивность заболевания высоковариабельна.
Риск рецидива. Низкий - в случаях новых мутаций, более высокий при половом мозаицизме и 50% - при наличии доминантного гена у родителей.
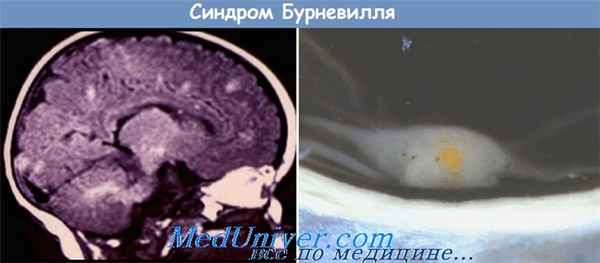
Диагностика. Диагноз обычно устанавливается на основании обнаружения опухолей сердца, которые напоминают небольшие фиброматозные узлы при миоме матки (в виде округлых образований однородной структуры с четкими контурами). От 51 до 86% рабдомиом сердца сочетается с туберозным склерозом. Иногда выявление рабдомиомы в ходе планового ультразвукового обследования плода во втором триместре беременности может привести к осознанию факта наличия болезни у матери. Рабдомиомы сердца имеют тенденцию к росту в пренатальном периоде и могут регрессировать в раннем неонатальном или оставаться неизменными в детском периоде и регрессировать в подростковом возрасте. Рабдомиомы могут вызывать нарушения сердечного ритма (синдром Вольффа-Паркинсона-Уайта (Wolff-Parkinson-White), суправентрикулярную тахикардию и пароксизмальную аритмию), а также явления обструкции или регургитации. Ангиофибромы почек пренатально не определяются, хотя это может быть просто вопросом времени с точки зрения совершенствования ультразвукового оборудования. Недавно сообщалось о возможности выявления перивентрикулярных субэпиндимальных узелков.
Патогенез. Обнаружены аномалии гена, ответственного за возникновение патологического гамарти-на и туберина - протеинов, которые могут играть роль в метаболизме GTP.
Генетические нарушения. Существует два локуса для туберозного склероза: один в хромосоме 9q и другой, возможно, в хромосоме 11q.
Дифференциальный диагноз. Доминирующим пренатальным признаком является рабдомиома. Следует также учитывать другие опухоли сердца, такие как фиброма.
Прогноз. Если в результате наличия рабдомиомы сердца не возникает водянки плода, то прогноз зависит от других осложнений заболевания. Вследствие большой вариабельности проявлений точное прогнозирование состояния ребенка бывает затруднено, и заключение во многом зависит от состояния здоровья матери. Более того, новые генетические данные указывают на различный прогноз в отношении умственного развития в зависимости от локализации дефектного гена, что, в свою очередь, может повлиять на принятие решения о последующей беременности.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Туберозный склероз
С середины XIX века в Европе фиксировались случаи заболевания, при котором в организме возникало большое количество опухолей. Патологию называли болезнью Бурневиля, а также сальной аденомой Прингла – по фамилиям врачей, которые занимались ее изучением. В начале ХХ века знания о заболевании были обобщены, но только к концу столетия появились данные, позволяющие сделать шаг на пути к эффективному лечению. Это стало возможным, благодаря открытию генов, мутация которых вызывает нарушение, и изобретению новых методов диагностики. Современные медики называют эту болезнь туберозным склерозом или комплексом туберозного склероза - аутосомно-доминантным заболеванием (из группы факоматозов), которое сопровождается развитием множественных доброкачественных опухолей (гамартом) в различных органах.
Симптомы туберозного склероза
Уже давно при обследовании пациентов и вскрытии умерших обнаруживали небольшие плотные опухоли различной локализации. Факты накапливались, и выяснилось, что подобные новообразования могут возникнуть где угодно - как в коже или головном мозге, глазах, глазных нервах, так и в сердце, почках, легких, или органах пищеварительной системы. Потому и проявления болезни зависят от того, какой орган поражен.
Самые явные симптомы подразделяются на большие (первичные): ангиофибромы лица, наличие белых (депигментированных) пятен на коже; узелки – фибромы возле ногтей; шагреневая (шероховатая) поверхность кожи; множественные доброкачественные опухоли сетчатки; фиброзные бляшки на лбу и других частях тела; опухолевидное поражение легких; множественные сосудистые опухоли почек. И на малые (вторичные) симптомы: “вмятины” в эмали зубов; опухоли внутренних органов; гамартомы ротовой полости и в желудочно-кишечном тракте (например, ректальные полипы); снижение остроты зрения из-за поражения сетчатки глаза (ахроматия).
Многие факоматозы протекают со снижением уровня интеллекта. Сыпь в виде мелких розоватых пузырьков (ангиофибром), расположенных на носу и щеках симметрично (в форме бабочки), а также на подбородке в сочетании с эпилепсией и низкими интеллектуальными способностями называли триадой Фогта, но все три признака проявляются одновременно далеко не у всех заболевших.
Если у ребенка в период внутриутробного развития появились опухоли в сердце (рабдомиомы), в некоторых случаях они приводят к смерти еще до рождения, а могут и не вызывать серьезных нарушений сердечно-сосудистой системы. Интересно, что новообразования могут даже уменьшаться в размерах и бесследно исчезать, но это, как правило, происходит только до 6-летнего возраста.
Причины заболевания
Тот факт, что у некоторых больных родственники также страдали и умирали от опухолей, натолкнуло медиков на мысль о наследственной передачи болезни. Со временем были выявлены гены TSC1 и TSC2, которые у здоровых людей защищают организм от появления новообразований. То есть мутация лишает человека естественной защиты от образования опухолей.
Изменения в генах могут произойти случайно, то есть больной ребенок может родиться и в семье, в которой туберозным склерозом не болеют. В других случаях люди наследуют мутации от своих родителей.
Как проводится диагностика
Туберозный склероз диагностируется примерно у 1 человека из 30 000 населения, с этим диагнозом рождается 1 ребенок из 6 000 – 10 000 новорожденных. Сложность диагностики заболевания вызвана его клиническим полиморфизмом, поэтому и проводится на стыке дисциплин.
Кроме выявления больших и малых патогномоничных симптомов для диагностики комплекса туберозного склероза используются такие методы, как:
ультразвуковое исследование – УЗИ (выявление кист, фиброзов внутренних органов). Признаки болезни Бурневиля обнаруживаются даже у плода в утробе матери во второй половине беременности. Если при проведении УЗИ выявляются рабдомиомы сердца, это может быть одним из первых проявлений туберозного склероза.
магнитно-резонансная томография – МРТ и компьютерная томография (КТ), причем более эффективной считается МРТ, где патология мозга верифицируется более убедительно. При данной патологии обнаруживаются так называемые корковые туберы – выступы в сером веществе, которые в половине случаев кальцифицируются (в них накапливается кальций). Также могут развиваться узлы в желудочках мозга, аномалии строения белого вещества.
электроэнцефалография – ЭЭГ для подтверждения уровня поражения ЦНС;
Очень важен для диагностики данного заболевания генетический анализ. Он позволяет выявить мутации, которые провоцируют развитие опухолей в организме. Однако не у всех людей, имеющих клинические проявления туберозного склероза, такие мутации обнаруживаются.
Лечение болезни Бурневиля
Почти все известные методы лечения направлены на устранение симптомов, которые доставляют дискомфорт пациенту и угрожают его жизни. Однако с 2012 года начал использоваться препарат, который влияет на основное звено патогенеза, является ингибитором сигнального пути и уменьшает рост опухолей в ЦНС и почках.
Пациентам назначаются в виде симптоматической терапии противосудорожные средства, хотя они зачастую оказываются неэффективными (необходимо проверять эффективность каждого препарата).
Если в сердце образуются рабдомиомы, и они мешают нормальному кровотоку, их удаляют хирургическим путем. При наличии кисты или карциномы в почках может проводиться операция по удалению новообразования или трансплантация органов. Для поддержания нормальной работы мочевыделительной системы обязательно нужно контролировать артериальное давление.
Профилактика болезни и ее осложнений
Причина туберозного склероза – это мутации, они могут передаваться по наследству или спонтанно возникают у человека во внутриутробном периоде. Поэтому для профилактики развития такого заболевания у детей нужно быть осведомленным о наличии подобных мутаций у себя и своего партнера. Генетический анализ особенно необходим, если у близких родственников выявлена болезнь Бурневиля, или у пары уже есть больной ребенок. Вероятность рождения второго ребенка с такой же патологией составляет 2%, если мутация возникла спонтанно, и 50%, если мутация передалась по наследству. Во время беременности проводится скрининговое обследование, для выявления признаков болезни у плода.
Если у пациента диагностирован туберозный склероз, проводят профилактические обследования для выявления нарушений в организме на самых ранних стадиях их формирования. Например, назначается ЭЭГ для выявления признаков, характерных для возникновения судорог.
Приступы эпилепсии негативно влияют на мозг и провоцируют снижение интеллектуальных способностей. Чтобы предотвратить это или ослабить вредное влияние, нужно регулярно проводить нейропсихологическое обследование, по результатам которого можно корректировать лечение. Необходима профилактика всех возможных проявлений заболевания: прием препаратов, которые снижают риск кровотечения, развития психических нарушений.
Повысить качество жизни пациента можно только при регулярном наблюдении. Поэтому нужно вовремя обращаться к специалистам при малейшем подозрении на заболевание, самолечение недопустимо. Больные должны обязательно выполнять все назначения врача. Некоторые лекарства приходится принимать в течение всей жизни, однако современные препараты могут существенно снизить угрозы для жизни.
Литература:
Шнайдер Н.А., Максимова Ю.В., Максимов В.Н., Дмитренко Д.В., Шаповалова Е.А. Туберозный склероз (болезнь Бурневилля-Прингла): Учебное пособие с грифом УМО Министерства образования РФ для системы последипломного образования врачей. / Под ред. Н.А. Шнайдер, Ю.В. Максимовой. ― Красноярск, 2010. – 112 с.
Шнайдер Н.А. Туберозный склероз: дефиниция, особенности клинического течения // Международный неврологический журнал. - 2010. - № 2(32).- С.5-13.
Болезнь Бурневилля–Прингля
Описаны болезнь Бурневилля-Прингля (туберозный склероз), механизмы ее наследования, клинические проявления, диагностические критерии, подходы к лечению и прогноз для пациентов.
Bourneville's disease (tuberous sclerosis), mechanisms of its inheritance, clinical presentations, diagnostic criteria, approaches to treatment and forecast for patients were described.

Болезнь Бурневилля–Прингля (туберозный склероз) — гетерогенное, генетически детерминированное заболевание, характеризующееся гиперплазией производных экто- и мезодермы, поражением кожи, нервной системы и наличием доброкачественных опухолей (гамартом) в различных органах. В 1880 г. описание болезни опубликовано Д. М. Бурневиллем и в 1890 г. Дж. Дж. Принглом.
Болезнь Бурневилля–Прингля наследуется по аутосомно-доминантному типу, отличается варьирующей экспрессивностью и неполной пенетрантностью. Развитие болезни определяется сцеплением с локусами 9q34 (первого типа — TSC1), Ilql4—llq23 и 16р13.3 (второго типа — TSC2). Имеются данные о наличии мутации гена на 12-й хромосоме. Предполагается, что гамартин (кодируется TSC1) является белком, подавляющим рост опухолей, а туберин (кодируется TSC2) регулирует эндоцитоз. Возможен дефект в системе репарации ДНК, о чем свидетельствует повышенная чувствительность клеток к ионизирующей радиации. В 50–75% случаев заболевание может быть обусловлено новыми мутациями. Частота болезни Бурневилля–Прингля составляет 1:30 000 населения. Распространенность среди новорожденных варьирует до 5–7 случаев на 100 000 новорожденных [1, 2].
В ангиофибромах наблюдается разрастание соединительной ткани, пролиферация мелких сосудов, преимущественно капиллярного типа, расширение их просветов. Соединительнотканные невусы при болезни Бурневилля–Прингля представлены коллагеномами. Эпителий обычно без особенностей, но может быть изменен по типу эпидермального невуса. Дерма утолщена за счет гипертрофированных коллагеновых волокон [1].
Клинические симптомы болезни Бурневилля–Прингля появляются в первые годы жизни, но могут существовать с рождения. Процесс постепенно прогрессирует, особенно в период полового созревания. Кожа поражена в 96% случаев [3]. Кожные проявления болезни Бурневилля–Прингля представлены ангиофибромами и фиброматозными очагами на лице, околоногтевыми фибромами, шагреневыми бляшками, гипомеланотическими пятнами, пигментными пятнами цвета «кофе с молоком». В 1998 г. были приняты диагностические критерии заболевания (табл.) [4].
По приведенным диагностическим критериям несомненный диагноз болезни Бурневилля–Прингля ставится в случае двух или одного первичного признака и двух вторичных признаков. Возможный диагноз: один первичный признак и один вторичный признак. Предположительный диагноз: или один первичный признак, или два (и более) вторичных признака (табл.).
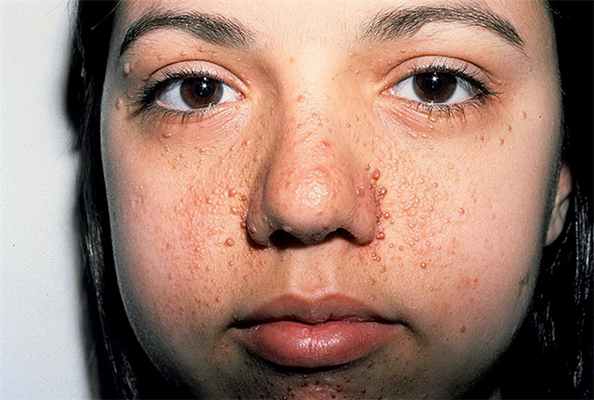
Гипопигментированные пятна на коже существуют с рождения или появляются в грудном возрасте и являются одним из наиболее частых кожных проявлений болезни Бурневилля–Прингля. На первом году жизни их находят у 80% больных, на втором — у 100% [3, 5]. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментные пятна при этом заболевании локализуются преимущественно на туловище и ягодицах. Характерной их особенностью является асимметричность расположения. Отмечена вариабельность числа, размера и формы пятен. Наиболее характерные из них имеют очертания листа, заостренного с одной стороны и закругленного с другой, бледно-сероватой или молочно-белой окраски. На светлой коже их видно только с помощью лампы Вуда. С течением времени пятна могут медленно репигментироваться. Диагностическое значение имеют только множественные элементы, особенно при сочетании их с эпилептиформными припадками. С младенчества могут выявляться белые пряди волос, ресниц и бровей, которые, как и гипопигментные пятна, являются характерным признаком болезни Бурневилля–Прингля.
Наряду с гипопигментными пятнами при болезни Бурневилля–Прингля в 15,4% случаев встречаются пигментные пятна цвета «кофе с молоком», которые не отличается от таковых у здоровых лиц, но наличие их в сочетании с другими симптомами помогает в постановке диагноза.
В 47–90% случаев наблюдаются ангиофибромы, являющиеся облигатным признаком болезни Бурневилля–Прингля. Ангиофибромы на первом году жизни появляются только у 20% больных, к трем годам — у 50%, располагаются, как правило, симметрично на крыльях носа, в носогубных складках и на подбородке. Они представляют собой мелкие полушаровидные плотноватые опухолевидные элементы, величиной 1–5 мм, телесного, желтовато-красного или красновато-коричневатого цвета. Их поверхность блестящая, гладкая, но может быть веррукозной, покрытой телеангиэктазиями (рис. 1, 2).
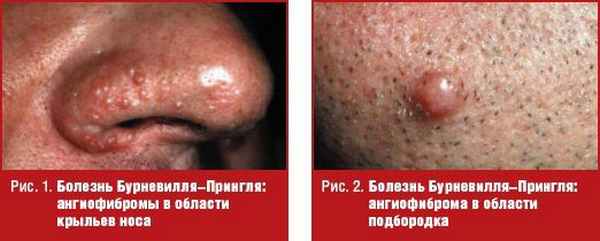
В области кожи лба, волосистой части головы, щек наблюдающиеся крупные опухолевидные фиброматозные очаги также являются облигатным признаком заболевания и встречаются у 25% больных болезнью Бурневилля–Прингля. Фиброматозные очаги могут быть как одиночными, так и множественными, имеют вариабельную окраску — от цвета нормальной кожи до светло-коричневого, несколько выступают над окружающей кожей, мягкой или плотноватой консистенции. Они часто появляются уже на первом году жизни и являются одним из первых клинических симптомов заболевания. Фиброзные бляшки чаще всего локализуются на лбу. Размеры и число их могут варьировать. Мягкие фибромы встречаются у 30% больных, представляют собой множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях. Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше 0,3 см в диаметре, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Часто встречаются подногтевые и околоногтевые фибромы (опухоли Кенена), гипертрофические изменения десен. Околоногтевые фибромы, являющиеся облигатным признаком болезни Бурневилля–Прингля, представляют собой тусклые, красные либо мясного цвета папулы или узлы, растущие от ногтевого ложа или вокруг ногтевой пластинки. Опухоль Кенена появляется в позднем детском возрасте и встречается в 17–52% случаев. В большинстве случаев околоногтевые фибромы появляются на втором десятилетии жизни. Наиболее часто они локализуются на ногах. Размер их варьирует от 1 мм до 1 см в диаметре. Гистологически опухоль Кенена представляет собой ангиофиброму.
Шагреневидные бляшки, или «шагреневая кожа», развиваются в первое десятилетие жизни примерно у 40% больных и представляют собой соединительнотканные невусы. В большинстве случаев «шагреневая кожа» появляется на втором десятилетии жизни. Шагреневидные бляшки могут быть как единичными, так и множественными, от мелкого размера до 10 и более см в диаметре, с пористой поверхностью типа «лимонной корки». Участки «шагреневой кожи» наблюдаются преимущественно в пояснично-крестцовой области, имеют вид плоских, слегка возвышающихся очагов, располагающихся преимущественно в люмбосакральной области, цвета нормальной кожи или слабо пигментированные.
При болезни Бурневилля–Прингля встречаются разнообразные системные изменения организма. Неврологические симптомы могут быть самыми первыми признаками болезни, внезапно проявляющимися на фоне внешнего здоровья и благополучия у ребенка без заметных дисплазий и нарушений развития. Возникают они в возрасте 3–4 месяцев в виде судорожных приступов. Поражения нервной системы являются доминирующими в клинической картине болезни Бурневилля–Прингля. Наиболее характерны судорожные пароксизмы, умственная отсталость, нарушения поведения, изменения в цикле «сон–бодрствование».
Судорожные пароксизмы — один из наиболее значимых симптомов болезни Бурневилля–Прингля — наблюдаются у 80–92% больных [6] и чаще всего являются манифестным симптомом заболевания. Первые приступы бывают общими тоническими, затем они становятся полиморфными (общие, фокальные, большие, малые, кивки, закатывание глаз, замирания, судороги). Частые припадки в основном наблюдаются до 6–7-летнего возраста, а затем они могут пройти. У ряда больных приступы продолжаются и в более старшем возрасте. У некоторых они имеют тяжелое течение, может развиться эпилептический статус с летальным исходом [7]. Чем раньше начинается эпилепсия, тем тяжелее умственная отсталость [3]. Эпилептические пароксизмы при болезни Бурневилля–Прингля нередко резистентны к противосудорожной терапии, могут приводить к развитию нарушений интеллекта и поведения и являются одной из главных причин инвалидности у детей с болезнью Бурневилля–Прингля. Среди факторов, детерминирующих резистентность к противосудорожной терапии, наибольшее значение имеют: дебют в возрасте до одного года, наличие нескольких типов приступов, высокая частота приступов, изменение характера приступов с течением заболевания [8].
Наиболее типичными поражениями головного мозга при болезни Бурневилля–Прингля являются корковые туберы, субэпендимальные узлы и аномалии белого вещества мозга. Кальцификация туберов отмечается в 54% случаев и увеличивается с возрастом больных. Большую значимость в верификации туберов при обследовании больных болезнью Бурневилля–Прингля имеет магнитно-резонансная томография (МРТ), которая позволяет визуализировать туберы в 95% случаев.
Субэпендимальные узлы встречаются в 95% случаев и выявляются как при компьютерном томографическом (КТ), так и при МРТ-исследованиях мозга. Субэпендимальные узлы в большинстве случаев множественные, прилегающие друг к другу. Локализуются, как правило, в стенках боковых желудочков, реже — в стенках III и IV желудочков мозга. Субэпендимальные узлы нередко трансформируются в гигантоклеточную астроцитому и выявляются у 10–15% больных [5]. Субэпендимальные гигантоклеточные астроцитомы манифестируют обычно между 5-м и 10-м годами жизни (средний возраст в момент выявления опухоли — 13 лет), как правило, имеют тенденцию к росту и всегда локализуются у межжелудочкового отверстия. У 10% больных при болезни Бурневилля–Прингля описаны поражения мозжечка.
Среди более редких неврологических симптомов встречаются центральные спастические параличи, пирамидные и экстрапирамидные симптомы, при росте опухоли в полость желудочков — внутренняя гидроцефалия. Могут быть обнаружены застойные соски зрительных нервов, их атрофия, в редких случаях — эндокринные расстройства в виде адреногенитального синдрома, нарушения со стороны черепно-мозговых нервов. В редких случаях наблюдаются спонгиобластомы, развивающиеся из очага болезни Бурневилля–Прингля, с соответствующей симптоматикой опухоли.
Развитие умственной отсталости замечается позже появления судорожного синдрома и регистрируется примерно у 49% больных, постепенно усугубляется вследствие деструкции мозга, пораженного болезнью Бурневилля–Прингля, и может достигать степени глубокой имбецильности. Отстает психическое развитие, разрушаются моторные навыки, нарушается речь. К пубертатному периоду нарушение интеллекта может достигать степени идиотии. Д. М. Бурневиллем (1880) описание этой болезни было опубликовано под названием «вклад в изучение идиотии». Однако при нерезко выраженном деструктивном процессе клинические проявления не столь тяжелы, эволютивная динамика развития нервной системы в известной степени перекрывает патологический процесс, и к психиатру этих детей приводят с олигофреническим интеллектуальным недоразвитием [3, 7].
Интеллектуальный дефект может резко углубляться при развитии психотических нарушений. Наблюдаются шизофреноподобные психозы со страхами, манией преследования, аномалии поведения с психопатическими чертами, изменениями личности по ограниченному типу с вязкостью, некритичностью, назойливостью. Даже при легких формах слабоумия, когда дефект нарастает медленно, годами, приходится помнить о том, что это заболевание имеет прогредиентное течение и, следовательно, неблагопрятный прогноз. Однако у 30% больных не отмечают слабоумия. Но статистика не точна, так как приводятся данные по регистрации обратившихся больных.
Нередко при поражении глаз выявляют застойные соски, иногда атрофию зрительных нервов. При офтальмоскопическом обследовании более чем у 50% больных наблюдается патогномоничная картина ретинальной факомы. Эти невоидные образования бывают трех типов. При первом, наиболее распространенном варианте гамартомы имеют нежную, относительно плоскую и гладкую поверхность, оранжево-розовый цвет, округлую или овальную форму, локализуются преимущественно в поверхностных слоях сетчатки. При втором — гамартомы имеют узловатый вид и напоминают тутовую ягоду. Они белого цвета, кальцифицированные, светонепроницаемые. При третьем варианте гамартомы сочетают в себе признаки первых двух. Они имеют округлую форму с узловатым и кальцифицированным центром и полупрозрачной, гладкой периферией оранжево-розового цвета. Такие проявления имеют важное диагностическое значение из-за характерного вида. У некоторых больных это может быть одним из единственных проявлений болезни Бурневилля–Прингля. Значительно реже регистрируются другие изменения органа зрения: хориоретинит, зоны депигментации, врожденная катаракта, врожденная слепота. Встречаются также соединительнотканные узелки на конъюнктиве. Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения [2, 7].
Опухоли во внутренних органах у многих больных не вызывают клинических симптомов, но часто обнаруживаются на аутопсии, особенно опухоли почек. Полагают, что опухоли почек выявляются у 40–50% больных. Это множественные билатериальные мелкие гамартомы из соединительнотканных волокон, жировой ткани, эпителия. Иногда встречаются крупные опухоли почек.
Изменения сердечно-сосудистой системы при болезни Бурневилля–Прингля проявляются развитием рабдомиом, которые нередко служат первым клиническим признаком болезни Бурневилля–Прингля наряду с гипопигментными пятнами. В 1863 г. Реклингхаузен описал сочетание поражения мозга с рабдомиомой. Рабдомиомы встречаются в 30–60% случаев и выявляются чаще у лиц мужского пола (соотношение 2:1). Наиболее высокая частота рабдомиомы сердца при болезни Бурневилля–Прингля наблюдается у новорожденных и детей грудного возраста. Рабдомиомы сердца, как правило, быстро увеличиваются во время второй половины беременности, в основном достигают максимальных величин к моменту рождения, а затем постепенно уменьшаются в размерах. Большинство рабдомиом исчезают бесследно. Спонтанная регрессия рабдомиом может быть у детей младше шести лет. После шести лет опухоли обычно не исчезают, однако могут несколько уменьшаться в размере. Регресс опухолей может наблюдаться как в размере, так и в их числе [5].
Не являются большой редкостью и поражения легких в виде фиброзных опухолей, фибролейомиом, кистозных образований, интерстициального фиброза. У больных возникают приступы диспноэ, возвратный спонтанный пневмоторакс, кровохаркание, легочная недостаточость. Описываются опухоли поджелудочной железы, печени, мочевого пузыря, желудочно-кишечного тракта и других органов. На слизистых оболочках встречаются фибромы десны, языка, глотки, гортани [7].
Дифференциальную диагностику при болезни Бурневилля–Прингля гипопигментированных пятен следует проводить с очаговой формой витилиго, анемическим невусом, отрубевидным лишаем, беспигментным невусом, послевоспалительной гипопигментацией. Ангиофиброму следует дифференцировать с трихолеммомой, сирингомой, внутридермальным невоклеточным невусом. Опухоль Кенена следует дифференцировать с простыми бородавками [3].
Прогноз для выздоровления неблагоприятный. При тяжелых системных изменениях высока летальность в детском и молодом возрасте от эпилептического статуса, сердечной, почечной или легочной недостаточности. Выраженность кожных изменений не влияет на риск вовлечения в процесс внутренних органов.
При лечении наиболее крупные элементы удаляют электрокоагуляцией, криодеструкцией, лазерным излучением. Наблюдается уменьшение размеров ангиофибром от Тигазона (по 1 мг на кг массы тела) [1]. Может быть полезна дермабразия, которую следует проводить после стабилизации процесса. Длительно назначают антиконвульсивные препараты (Дифенин и др.), периодически — средства, снижающие внутричерепное давление, нормализующие сердечный ритм при рабдомиоме сердца. Терапия выбора при опухолях головного мозга — хирургическое удаление. С целью пренатальной диагностики может быть использована эхокардиография для выявления у плода рабдомиомы сердца.
Литература
- Мордовцев В. Н., Мордовцева В. В., Мордовцева В. В. Наследственные болезни и пороки развития кожи. Атлас. М.: Наука, 2004. С. 40–42.
- Страхова О. С., Катышева О. В., Дорофеева М. Ю., Перминов В. С., Пивоварова А. М., Осипова Э. К., Добрынина М. В., Чумак О. И. Туберозный склероз // Российский медицинский журнал. 2004. № 3. С. 52–54.
- Фицпатрик Т., Джонсон Р., Вулф К., Полано М., Сюрмонд Д. Дерматология: атлас-справочник. 1999. С. 460–466.
- Roach E. S., DiMario F. J., Kandt R. S., Northrup H. Tuberous Sclerosis Consensus Conference: Recommendations for Diagnostic Evaluation // Journal of Child Neurology. 1999. V. 14. Р. 401–407.
- Дорофеева М. Ю. Туберозный склероз у детей // Российский вестник перинатологии и педиатрии. 2001. № 4. С. 33–41.
- Curatolo P., Seri S. Seizures. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003. Р. 46–77.
- Суворова К. Н., Куклин В. Т., Рукавишникова В. М. Детская дерматовенерология. Казань, 1996. С. 50–56.
- Curatolo P. Tuberous Sclerosis. In: Infantile Spasms and West Syndrome. Ed. by O. Dulac, H. T. Chugani, B. Dalla Bernandina. W. B. Saunders. Company Ltd, London, Philadelphia, Toronto, Sydney, Tokio. 1994. Р. 192–202.
Л. А. Юсупова, доктор медицинских наук
З. Ш. Гараева, кандидат медицинских наук, доцент
Е. И. Юнусова, кандидат медицинских наук, доцент
Г. И. Мавлютова, кандидат медицинских наук
Л. А. Хаертдинова, кандидат медицинских наук, доцент
Э. Э. Галиханова, кандидат медицинских наук
В. Н. Рокицкая, кандидат медицинских наук, доцент
ГБОУ ДПО КГМА Минздравсоцразвития России, Казань
Что такое туберозный склероз: симптомы, диагностика и лечение
Туберозный склероз — редкое (1 случай на 30 000 человек) генетическое заболевание, вызывающее образование опухолей в органах по всему телу. Поражаются чаще всего внутренние органы (почки, сердце, легкие), но на начальной стадии заболевание может проявлять себя внешне: на коже лица появляются множественные новообразования.
Впервые «туберсы» (от лат. tuber — нарост) — новообразования в головном мозге — описывает невролог Бурневилль, поэтому в некоторых источниках данную болезнь можно встретить под его именем.
Лечение и прогноз туберозного склероза — задача не из легких: здесь нужен целый ряд грамотных специалистов и проведение множества обследований. При этом прогноз будет зависеть от здоровья пациента в целом, как быстро ему поставили правильный диагноз и как скоро начали лечение. В лучшем случае пациенты с ТС могут прожить довольно долгую жизнь.

Причины туберозного склероза
Болезнь Бурневилля имеет генетическую природу и обусловлена мутациями в генах TSC1 и TSC2, в связи с чем у больных наблюдается неконтролируемый рост опухолевых тканей.
Выделяют 2 типа туберозного склероза в зависимости от локализации:
- Мутация гена 34 локуса девятой хромосомы. В этом случае происходит нарушение кодирования гематина — антионкогена, обеспечивающего профилактику опухолевой трансформации клеток;
- Мутация в 13 участке шестнадцатой хромосомы приводит к сбоям в работе туберина — белка, блокирующего неконтролируемый рост клеток.
Мнение эксперта
Автор: Андрей Игоревич Волков
Врач-невролог, кандидат медицинских наук
Туберозный склероз — очень редкое заболевание, генетическое, характеризуется развитием доброкачественных опухолей в различных органах. ТС диагностируется в период полового созревания. Примерная частота — 1 на 6000. Если один из родителей имеет такое заболевание, то шанс того, что оно обнаружится у ребенка, составляет 50%. В то же время есть статистика, показывающая что 2/3 случаев — это новые мутации.
Клинические проявления заболевания очень разнообразны и зависят от локализации опухолей. Поражения ЦНС перекрывают поток нервных импульсов, приводят к задержке когнитивного развития, провоцируют судороги, спазмы. Туберомы могут разрастаться и перекрывать поток спинномозговой жидкости, что становится причиной односторонней гидроцефалии. Ангиолипомы (опухоли почек) и поликистоз почек провоцируют развитие артериальной гипертензии. Очень часто встречаются поражения кожи.
Для диагностики используются большие и малые признаки. Чтобы поставить диагноз туберозный склероз, достаточно двух больших признаков или одного большого признака в сочетании с двумя (и более) малыми. Обязательно нужны анализы крови и мочи, УЗД почек, ЭКГ, ЭЭГ, КТ, МРТ, генетические исследования.
Формы заболевания
ТС имеет два основные формы:
- Семейная — обнаруживается в семьях, где был больной родственник. У ребенка, чей родитель болен ТС, шанс заболевания — до 50%.
- Спорадическая — не носит наследственный характер, возникает спонтанно.
Симптомы туберозного склероза
Болезнь Бурневилля имеет довольно вариабельную клиническую картину, но в целом все признаки протекания болезни можно разделить на первичные и вторичные.
- ангиофибромы (новообразования с волокнистым и сосудистым типом ткани) лица или фиброзные бляшки (плотные округлые белые образования) на лбу;
- новообразования возле ногтей;
- нарушение пигментации кожи: появление трех и более гипопигментных пятен;
- выраженная сухость кожных покровов, так называемая шагреневая кожа;
- множественные новообразования на сетчатке (гемартомы);
- туберсы на головном мозге на границе белого и серого вещества;
- опухоль ЦНС;
- гиганто-клеточная астроцитома;
- рабдомиома (узел) сердца;
- кистозная деструкция легочной ткани;
- множественные новообразования на почках.
- многочисленные углубления в эмали зубов;
- полипы в кишечнике;
- разрушение костной ткани (костные кисты);
- фибромы полости рта;
- доброкачественные опухоли (гамартомы) внутренних органов;
- изменение цвета сетчатки глаза;
- нарушение пигментации кожных покровов;
- кисты в почках.
Для точного диагностирования туберозного склероза специалисту нужно подтверждение, как минимум двух первичных признаков или одного первичного признака и двух вторичных признаков.
У детей симптоматика следующая:
- головокружение и замедленная речь;
- временное ухудшение зрения, боль в глазах;
- понижение мышечного тонуса;
- дрожь конечностей (при поражении мозжечка);
- недержание мочи (при сложном протекании болезни);
- тошнота и рвота.
Поражение ЦНС
Поражение центральной нервной системы может проявляться по-разному. У пациента могут наблюдаться:
- приступы судорог (встречаются уже на первом году жизни ребенка, больного туберозным склерозом);
- изменение поведенческих реакций (аутизм, синдром гиперподвижности и дефицита внимания, агрессия и аутоагрессия);
- нарушение интеллекта (умственная отсталость) — отмечается в 50% случаев (варьируется от умеренной до глубокой);
- нарушение сна (бессонницы, снохождение, раннее пробуждение и пр.).
При поражении ЦНС на мозге появляются:
- Корковые туберсы (единичные и множественные) — выступы над бороздами головного мозга. Наблюдаются у больных в 50% случаев;
- Субэпендимарные узлы локализуются в желудочках головного мозга. В 10-15% случаев могут перерасти в гигантоклеточные астроцитомы. В этом случае у пациентов наблюдаются сильная головная боль, тошнота и рвота.
Дерматологические симптомы
Данное заболевание довольно активно проявляет себя внешне. Так, у больного могут обнаруживаться следующие симптомы:
- гипопигментные пятна — белые пятна на теле и ягодицах. Часто обнаруживаются с рождения, с возрастом их число растет;
- ангиофибромы лица — розоватые узлы на щеках, носу и подбородке;
- «шагреневая кожа» — желтоватая кожа с характерной поверхностью (похожа на цедру апельсина), локализуется обычно в пояснично-крестцовом отделе;
- околоногтевые фибромы — красноватые узлы вокруг ногтя, обычно на ногах.
- фиброзные бляшки — новообразования цвета кожи, немного шероховатые на ощупь; локализуются обычно на лбу;
- белые пряди волос, ресниц и бровей (часто данный симптом списывается родителями на детский возраст, и ему не придается большого значения. Но если белые волосы сочетаются с другими симптомами ТС, стоит посетить врача).
Офтальмологические симптомы
У 50% пациентов наблюдаются офтальмологические симптомы, сложность заключается лишь в том, что их трудно обнаружить на ранних этапах. Проявляется заболевание на этом уровне в появлении доброкачественной опухоли (гамартома, нередко — множественных) сетчатки и зрительного нерва. Обычно опухоль локализуется рядом с оптическим диском.
Проявляет себя гамартома падением зрения, депигментацией радужки, отеком диска зрительного нерва, косоглазием, катарактой и пр.
Поражение внутренних органов
Новообразования внутренних органов при ТС часто бывают множественными и поражают парные органы. Какое-то время течение болезни может быть бессимптомным. Чаще других при ТС поражаются следующие органы:
- сердце (рабдомиома) — при внутриутробном развитии рабдомиомы — вероятна гибель плода. У маленьких детей о наличии рабдомиомы могут свидетельствовать тахикардия, аритмия, фибрилляция желудочков. У детей постарше симптомов вообще может не быть, а к 5-6-летнему возрасту рабдомиома может исчезнуть совсем);
- легкие (кисты) поражаются уже в сознательном возрасте, обычно подростковом, приводит к дыхательной недостаточности;
- почки (поликистоз) — симптом характерен для половины больных и выступает вторым, приводящим к смерти (после поражения ЦНС). Данная проблема обнаруживается обычно у больных после 30 лет;
- печень (гамартома — доброкачественная опухоль, которая сопровождается одышкой, рвотой, дыхательной дисфункцией);
- кишечник (полипы — доброкачественные новообразования на ножке, свисающие со стенок органа. Возможны приступы схваткообразных болей и кровотечение).
Два последних нарушения приводят также к образованию дефектов зубов (углубления на эмали).
Диагностика туберозного склероза
ТС довольно трудно диагностировать. Как уже говорилось выше, иногда заболевание может протекать бессимптомно. Так как болезнь проявляет себя в разных органах (от глаз до кишечника), необходимы будут совместные усилия нескольких специалистов и проведение обширного обследования. Для начала специалистами проводится опрос пациента, который обычно содержит следующие вопросы:
- Какие симптомы?
- Как давно они появились?
- С какой частотой повторяются?
- Были ли в семье случаи данного заболевания?
Невролог осматривает пациента с целью найти какие-либо неврологические патологии (приступы эпилепсии, расстройства интеллекта и пр.).
Дерматологический осмотр нацелен на нахождение бугорков на коже или признаков ее обесцвечивания.
Офтальмолог осматривает глазное дно, сетчатку, зрительный нерв.
Также проводятся следующие обследования:
- Анализ крови на наличие повышенного уровня креатина, мочевины (наблюдается при поражении почек);
- Анализ мочи на наличие крови в ней (также симптом нарушения работы почек);
- Эхокардиография призвана показать бугорки на сердце;
- ЭЭГ с пробами поможет зарегистрировать церебральную эпилептическую активность.
- Чтобы оценить силу поражения ЦНС необходимо провести КТ и МРТ головного мозга. В случае туберозного склероза здесь будут видны бугорки на поверхности головного мозга, а также увеличение количества ликвора.
- При подозрении на локализацию ТС в конкретном органе проводится УЗИ, КТ и МРТ этого органа или совокупности (например, УЗИ брюшной полости), при подозрении поражения кишечника проводится ректороманоскопия и колоноскопия. Диагностика офтальмологических поражений происходит путем проведения офтальмоскопии (осмотр глазного дна с целью оценки состояния сетчатки).
- Генетическое обследование на наличие изменений в генах TSC1 и TSC2.
Лечение туберозного склероза
Полностью вылечиться от ТС на данный момент невозможно, так как заболевание является генетическим. Курс терапии призван прервать признаки болезни и улучшить общее состояние пациента.
Антиконвульсантная терапия является основной в лечении ТС, так как данное заболевание сопровождается частыми припадками эпилепсии, а степень олигофрении напрямую связана с частотой эпиприступов.
Врачами обычно назначаются препараты следующего рода:
- Гипотензивные — призваны понизить давление;
- Сердечно-сосудистые — в случае нарушения работы сердца;
- Кортикостероиды, противосудорожные средства и антидепрессанты — для ликвидации приступов эпилепсии и пр.
Пациентам с ТС также важно правильно питаться: необходимо увеличить потребление жиров и уменьшить — белков и углеводов.
В случае тяжелого протекания болезни пациентам назначается операция (в зависимости от места локализации опухоли): удаляют опухоли в головном мозге, почках, органах ЖКТ и пр. Если опухоли множественные, нередко удаляется орган целиком. Бугорки на коже удаляют лазером или методом криодеструкции.
Для детей также необходима консультация психотерапевта для предупреждения олигофрении. Лечение в детском возрасте призвано устранить нервные судороги. При локализации опухолей в сердце или почках лечат сердечную и почечную недостаточность. Внутричерепные опухоли подлежат удалению, так как они провоцируют рост внутричерепного давления. Вовремя поставленный диагноз и грамотное лечение дает шансы ребенку прожить довольно долгую жизнь.
Осложнения
ТС — заболевание довольно опасное: диагностировать его трудно, вылечить практически невозможно, так еще и встречается ряд осложнений, а именно:
- Гемианопсия — поражение зрительной системы. Человек с таким диагнозом видит мир будто наполовину;
- Водянка головного мозга (гидроцефалия) опасна сильными головными болями, снижением слуха и зрения;
- Почечная недостаточность ведет к задержке шлаков в организме, нарушению кислотно-щелочного баланса, сбоям в работе сердца;
- Стойкая эпилепсия — это невозможность для пациента прийти в сознание даже на время между приступами. Эпилептический статус (затянувшийся приступ) может привести к летальному исходу.
ТС опасен целым рядом осложнений, каждое из которых, в свою очередь, требует отдельного лечения, именно поэтому так важно вовремя поставить правильный диагноз и приступить к лечению ТС.
Прогноз заболевания
Полностью данное заболевание устранить невозможно, но вот облегчить симптомы — вполне. Поэтому так важно диагностировать ТС на ранних этапах: возможно для борьбы с признаками еще будет достаточно традиционной терапии без хирургического вмешательства.
Поэтому при обнаружении ряда вышеописанных симптомов (особенно у детей) сразу необходимо проконсультироваться с врачом, сдать необходимые анализы и пройти обследование. Так как ТС — нарушение генетическое, то людям, в чьих семьях оно встречалось, на стадии планирования беременности рекомендуется консультация медицинского генетика.
Туберозный склероз
Туберозный склероз, или заболевание Бурневилля, является редким генетическим нарушением, при котором отмечается склонность к развитию доброкачественных опухолей по всему организму. Показатели заболеваемости составляют один случай на 30 тыс. населения. У новорожденных этот показатель ниже в 3–4 раза (один случай на 6–10 тыс.). Хотя туберозный склероз является наследственным заболеванием, существует вероятность возникновения спорадических случаев.
Почему возникает туберозный склероз?
Наследование заболевания осуществляется по аутосомно-доминантному типу и связано с генами, распложенными в девятой (длинное плечо, участок 34) и тринадцатой хромосоме (короткое плечо, участок 13). В первом случае нарушается кодирование белка гамартрина, а во втором ― туберина. Точная роль этих белков в организме до конца не изучена, однако известно, что они участвуют в механизмах, ответственных за подавление роста опухолевых клеток. При нарушении функций данных белков оказываемый ими эффект изменяется в противоположную сторону, что и приводит к развитию опухолей.
Клинические проявления туберозного склероза
В зависимости от того, в каком органе локализован патологический очаг, у пациента может наблюдаться разнообразная клиническая картина. Поражение головного и спинного мозга при туберозном склерозе являются самыми частыми и сопровождаются следующей симптоматикой:
- судорожный синдром;
- олигофрения;
- спазмы отдельных групп мышц;
- задержка нейропсихического развития;
- эмоциональная нестабильность;
- нарушения сна.
Практически в каждом случае туберозного склероза отмечаются различные поражения кожи. Данная группа симптомов отличается большим разнообразием, но среди наиболее частых признаков можно выделить:
- очаги гипер- и гипопигментации;
- опухоль из сосудов и соединительной ткани (ангиофиброма), локализованная в области лица;
- фиброзные бляшки;
- дерматофиброма;
- околоногтевая фиброма.
Среди органов-мишеней при туберозном склерозе можно отметить легкие, почки, сердце, кишечник, печень. В зависимости от объема поражения и конкретного органа у пациента проявляются специфические симптомы, например при поликистозе почек отмечается артериальная гипертензия, гематурия, полиурия и др. Выраженная клиническая картина проявляется не всегда. Часто встречаются формы туберозного склероза, для которых характерно длительное бессимптомное течение, что сильно затрудняет своевременную диагностику заболевания.

Подход к диагностике
Диагностика туберозного склероза возможна лишь при комплексном обследовании пациента. При этом необходимо подключить большое количество специалистов (нефролога, офтальмолога, невролога, кардиолога, дерматолога и т. д.). Список обследования может включать в себя такие методы, как электроэнцефалография, МРТ, КТ, УЗИ, нейросонографию, осмотр глазного дна, томографию сетчатки и др.
Существует и более точная, генетическая диагностика туберозного склероза. Суть её заключается в поиске мутаций в генах TSC1 и TSC2 методом секвенирования, с помощью которого можно получить аминокислотную последовательность, характерную для того или иного белка, и сравнить её с вариантом нормы. Секвенирование является точным методом диагностики и отлично подходит для выявления туберозного склероза на доклиническом этапе. Пройти обследование рекомендуется всем лицам, у которых имеются случаи заболевания у близких родственников. Тест на туберозный склероз также могут сдать люди, у которых имеются характерные признаки болезни. Кроме того, диагностику может пройти любой желающий, для этого требуется сдать кровь из вены.
Таргетная терапия эпилепсии
«Эверолимус» — эффективный, приемлемо переносимый препарат в лечении субэпендимальных гигантоклеточных астроцитом и ангиомиолипом, ассоциированных с туберозным склерозом. Он обладает потенциалом таргетного воздействия на множество клинических проявлений туберозного склероза (в том числе эпилепсию), снижая риск жизнеугрожающих осложнений.
Для лечения инфантильных спазмов препаратами первого выбора являются «Вигабатрин» («Сабрил», «Сабрилекс») и гормональные средства (синактен депо — синтетический полипептид со свойствами эндогенного адренокортикотропного гормона).
В медико-генетическом центре «Геномед» выполняется широкий спектр молекулярно-генетических исследований, в том числе и анализ на туберозный склероз. С помощью современного оборудования и квалифицированного персонала можно получить объективную и точную информацию о состоянии здоровья, выявить широкий спектр генетических нарушений, после чего подобрать адекватную тактику лечения.
Читайте также:
- Неотложная помощь при остром коронарном синдроме.
- Физиология инсулина. Воздействие инсулина на клетку
- Инфраклинический гипотиреоз. Психоневрозы микседемы
- Решетчатый лабиринт Топография решетчатого лабиринта Слизистая околоносовых пазух Кровоснабжение околоносовых пазух Иннервация околоносовых пазух
- Неспецифическая интерстициальная пневмония