Вариабельная протеаза-чувствительная прионопатия
Добавил пользователь Alex Обновлено: 07.01.2026
ПРИОНЫ И ИХ РОЛЬ В РАЗВИТИИ
НЕЙРОДЕГЕНЕРАТИВНЫХ
ЗАБОЛЕВАНИЙ
Выполнили: студенты 2 курса 4 группы
л.ф. Низов А.
Дьяков Г.
Тагаев Ф.
студентка 2 курса 3 группы л.ф.
Попихина Д.
Научный руководитель: к.с/х.н., доцент
Гусева Т. М.
ОПРЕДЕЛЕНИЕ ПРИОНОВ
Прионы — уникальный класс
инфекционных агентов,
вызывающих «медленные
болезни»
Основные характерные черты
этой особой формы инфекции:
поражение одного органа или
одной системы и наличие
одного хозяина
продолжительный
инкубационный период
нарастание клинической
симптоматики, неизбежно
приводящее к смерти.
БИОХИМИЧЕСКАЯ ХАРАКТЕРИСТИКА
ПРИОНОВ
Прионы — инфекционные белки с низкой
молекулярной массой, не имеют нуклеиновых
кислот, не вызывают воспаления и иммунного
ответа, устойчивы к высокой температуре,
формальдегиду, к различным видам излучений,
нечувствительны к интерферонам и не индуцируют
их синтез
БИОХИМИЧЕСКИЕ ОСНОВЫ ПАТОГЕНЕЗА
ПРИОНОВ
Как правило, при переходе
белка в прионное состояние
его α-спирали
превращаются в β-слои.
Появившиеся в результате
такого перехода прионы
могут в свою очередь
перестраивать новые
молекулы белка; таким
образом, запускается цепная
реакция, в ходе которой
образуется огромное
количество неправильно
свёрнутых молекул.
Все известные прионы
вызывают формирование
амилоидов — белковых
агрегатов, включающих
плотно упакованные β-слои.
ОСНОВНЫЕ ПРИОННЫЕ ЗАБОЛЕВАНИЯ
Заболевание
Краткая характеристика
Вариабельная протеазачувствительная прионопатия
редкое ПЗ, диагностируется в 2—3 случаях
на 100 млн населения. Сходна с болезнью
Герстмана—Штраусслера—Шейнкера
Фатальная бессонница
редкое ПЗ, вызывающее нарушения
сна, двигательные расстройства и
приводящее к летальному исходу.
Куру
редкое ПЗ, эндемичное для
высокогорных районов Папуа — Новой
Гвинеи, распространялось через
ритуальный каннибализм.
БОЛЕЗНЬ КРЕЙТЦФЕЛЬДТА—ЯКОБА
Наиболее распространенное ПЗ
человека, на долю которого
приходится около 85% всех
случаев ПЗ
Нейродегенеративный процесс
может запускаеться спонтанно в
результате соматической генной
мутации, может передаваться
путем трансплантации роговицы
или при применении гормона
роста, приготовленного из
гипофиза человека, после
употребления в пищу говядины,
зараженной прионами
БОЛЕЗНЬ ГЕРСТМАНА—ШТРАУССЛЕРА—
ШЕЙНКЕРА
Подострая губкообразная энцефалопатия,
наследуется по аутосомно-доминантному типу
и обычно развивается в среднем возрасте.
Болезнь начинается с развития мозжечковой
атаксии с трудностями при ходьбе и
удерживании равновесия, к которым
присоединяются прогрессирующие изменения
личности и деменция.
ЗАКЛЮЧЕНИЕ
Изучение прионов и вызываемых ими
заболеваний является сравнительно
новой и быстро развивающейся
областью биомедицинских
исследований. В настоящее время
медицина не располагает
эффективными средствами лечения
данной патологии, поэтому важное
значение имеет профилактика
заражений прионами как
алиментарным (употребление
зараженных мясных продуктов), так
и трансмиссивным (применение
лекарственных биопрепаратов,
получаемых из тканей животных)
путями.
Прионные заболевания человека: симптоматика и перспективы лечения
Механизмы человеческих заболеваний, вызываемых прионами (англ. от Protein («белок») и Infection («Инфекция»); впервые слово было использовано С. Прузинером [1] в конце XX века), до сих пор остаются малоизученными, несмотря на, казалось бы, большой объём исследований, проводимых в данной сфере. Цель настоящей статьи заключается в обобщении и понятном объяснении имеющейся на данной момент информации, касающейся прионов и ассоциированных с ними болезней.
В рамках настоящей статьи приняты следующие сокращения: prion — proteinacious infectious particle; PrP — прионный белок; PrPC — нормальная изоформа прионного белка; PrPSc — инфекционная форма прионного белка; PRNP — ген, кодирующий прионный белок.
Прионные заболевания (также известны как трансмиссивные губчатые энцефалопатии, ТГЭ), ставшие известными человеку в середине XVIII века, являются одним из наиболее интригующих биологических феноменов. Исследования этого явления начались в XX веке с попыток определить биологическую сущность возбудителей сразу нескольких специфичных болезней животных и человека со схожей симптоматикой. Гипотеза об их общей этиологии, выдвинутая в 1960-х годах учёными радиобиологом Т. Альпером и математиком Д. Гриффитом [2] и позже дополненная и доказанная врачом С. Прузинером [3], дала толчок последующим исследованиям в этой области. Однако, несмотря на глубокую заинтересованность научного мира, многие аспекты существования прионов остаются неизученными и по сей день.
К числу человеческих болезней, связанных с этими особыми белковыми инфекционными агентами, относятся болезнь Якоба-Крейтцфельда (CJD) и её различные вариации, фатальная бессоница (FFI/FSI), болезнь Герстмана-Шраусслера-Шейнкера (GSS), куру, вариабельная протеаза-чувствительная прионопатия, и прионное заболевание, связанное с диареей и поражением вегетативной нервной системы.
Все вышеперечисленные прионные болезни на сегодняшний день остаются смертельными, что помещает их в категорию наиболее опасных болезней.
Сущность прионов
После окончания процесса перевода генетической информации, заключённой в виде нуклеотидной последовательности РНК (рибонуклеиновой кислоты), в специфическую последовательность аминокислот, формирующую первичную структуру всех протеинов, новосинтезированные белки сворачиваются в определённые структуры. Прионы — разновидность белковых молекул с неправильной «укладкой» (Рис. 1), дефектная форма нормального мембранного белка PrP, который экспрессируется (проявляется) преимущественно в клетках центральной нервной системы.
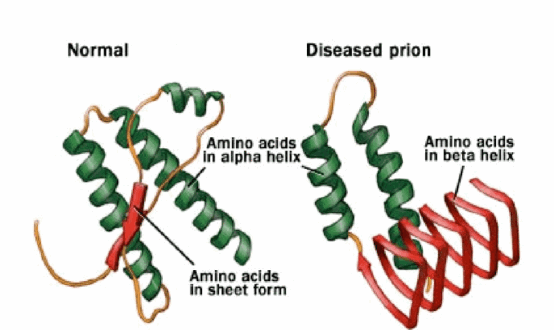
Рис. 1. Правильно «сложенный» (слева) и дефектный (справа) белки [4]
Благодаря определённым биологическим машинериям, обычные неправильно сложенные белки легко утилизируются и не оказывают отрицательного эффекта на процессы человеческой жизнедеятельности. Прионы отличаются устойчивостью к этим механизмам и наличием способности превращать нормальные составляющие их белки в себе подобные. Существует две гипотезы, описывающие механизмы этого явления. Согласно первой, гетеродимерной модели (Рис. 2) [5], превращение происходит следующим образом: PrPSc присоединяется к «здоровой» молекуле PrP, и катализирует ряд конформационных изменений, приводящих к её переходу в прионную форму, после чего уже два ненормальных белка расходятся и запускают новые раунды этого процесса. При этом наличие агрегированной («склеенной») формы белка не является обязательной частью прионной трансформации.
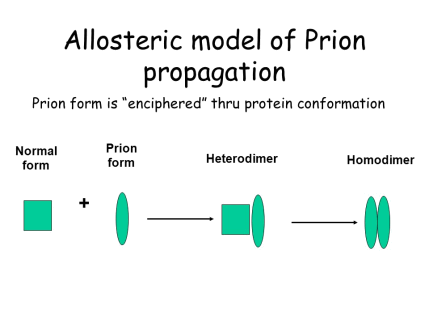
Рис. 2. Гетеродимерная модель прионной репликации [6]
Альтернативная гипотеза (Рис. 3) [5] — полимеризационная — гласит, что катализ конформационного превращения нормального белка в патологический может происходить только при «нуклеализации» с последующим образование олигомерных или мультимерных комплексов. Стоит отметить, что последние исследования говорят в пользу второй модели.
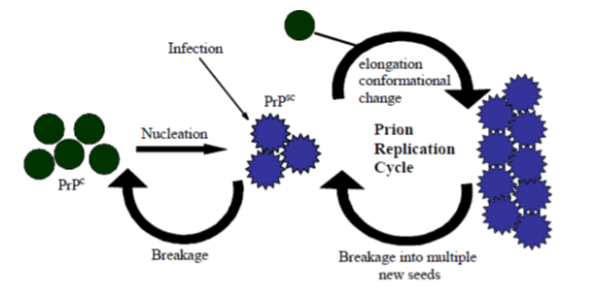
Рис. 3. Цикл репликации PrPSc в соответствии с полимеризационной гипотезой [7]
Воздействие PrPSc приводит к этакой «внутриклеточной эпидемии»: образуется множество нефункциональных белковых бляшек на клетках организма, из-за чего он рано или поздно погибает.
Пути возникновения прионных заболеваний
Считается, что существует всего три пути приобретения прионных заболеваний: прямое инфицирование, наследственная передача и спорадическое возникновение по неизвестному механизму [8], но вне зависимости от происхождения они могут быть переданы инфекционным путём.
Был детектирован высоконсервативный ген PRNP, несущий информацию о нормальной изоформе белка PrP, находящийся в p-плече 20-ой хромосомы человека [9]. PRNP имеет протяженность 16 тысяч нуклеотидных последовательностей и содержит 2 экзона. Все наследственные прионные заболевания связаны с аутосомным наследованием мутаций, произошедших в данном гене.
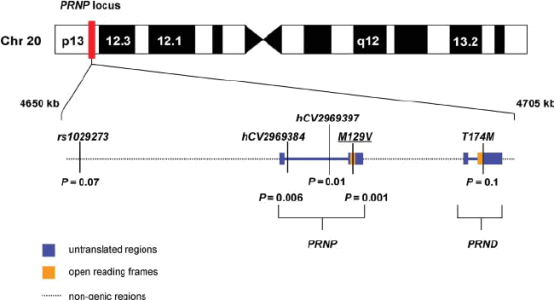
Рис. 4. Локализация гена PRNP в 20-ой хромосоме человека [10].
Основной способ возникновения прионной болезни — спонтанный. Согласно одной из гипотез, объясняющих этот процесс, в нормальных белках происходит определённая посстрансляционная модификация [11]. Иная гипотеза постулирует, что в какой-то конкретный момент неопределённое количество клеток организма соматически (ненаследственно) мутирует и начинает производить дефектный белок PrPSc [12].
Попасть в незаражённый человеческий организм прион может различными путями: при поедании плохоприготовленного мяса, содержащего PrPSc, переливании крови от инфицированного человека к здоровому, трансплантации заражённых органов и тканей.
Клиническая картина прионных заболеваний человека
Ещё одной важной особенностью прионных белков является наличие возможности принимать определённое число различных конформаций. Это обуславливает отличия в течении и симптоматике прионных заболеваний: возможны различные инкубационные периоды, повреждения разных участков коры головного мозга, нарушения различных функций нервной системы [13]. Несмотря на это, среди всех ассоциированных с действием прионов заболеваний прослеживается серия общих чёрт: поражение нервной системы, изначальное отсутствие иммунного ответа на дефектные белки PrP вследствие постоянного присутствия их «правильной» изоформы в организме [5], быстрая прогрессия болезни после окончания инкубационного периода.
Болезнь Крейтцфельда-Якоба (CJD)— редкое, но тем не менее наиболее известное прионное заболевание человека. Существует несколько форм CJD — наследственная, ятрогенная, спорадическая, вариативная, при этом первые три отличаются преимущественно способом распространения. Патоморфологические картины наследственной, ятрогенной и спорадической CJD схожи: во всех случаях наблюдаются прогрессирующие когнитивные нарушения, поражения и дисфункуция мозжечка или комбинация этих расстройств; нарушение зрения вплоть до слепоты; миоклонические припадки. В терминальной стадии появляются глобальные когнитивные нарушения, смерть наступает через 8–10 месяцев после диагностирования CJD [14]. Вариативная CJD имеет несколько более глубоких отличительных особенностей: поражает молодых людей в возрасте в среднем до 30 лет, ее начало характеризуется изменениями поведения, бессонницей, депрессией; двигательные нарушения проявляются примерно через 6 месяцев от начала заболевания в виде прогрессирующей атаксии, хореи, миоклонуса; слабоумие наступает позднее, чем при классической форме, пациент осознает свое ухудшающееся состояние. Для вариабельной БКЯ типичны не только начало в более молодом возрасте, но и средняя выживаемость, превышающая 14 месяцев [15].
Болезнь Герстмана-Штраусслера-Шейнкера (GSS)— заболевание, несколько отличающееся от CJD по нескольким признакам. Эта клиническая форма ТГЭ вызвана мутацией гена PNRP в 102-ом кодоне, приводящей к замене аминокислоты пролин на лейцин [16]. Болезнь начинается в среднем возрасте с проявления мозжечковой атаксии, речевых расстройств, деменции и измнений в поведении. К числу этих симптомов могут прибавляться диплопия, глухота, миоклонические приступы, спастичность.
Куру— прионное заболевание, эндемичное для некоторых районов Папуа-Новой Гвинеи. Основным способом распространения этой болезни был ритуальный каннибализм. Симптоматика включает в себя двигательные расстройства (тремор, массивные фасцикуляции, хореоатетоз, миоклонии). Смерть наступает через приблизительно 2 года после дебюта заболевания. В 2009 году было уставлено, что некоторые члены одного из аборигенных племён обладают врождённым иммунитетом к куру благодаря появлению у них относительно новой полиморфической модификации гена PNRP [17].
Фатальная бессоница — редкое ТГЭ, обычно связанная с наследованием аутосомно-доминантной мутации («семейная», FFI). Отмечается, что существует и спорадическая форма этого заболевания («спонтанная», FSI) [18]. В обоих случаях наблюдается следующая картина: нарушения сна, галлюцинации, вегетативная гиперактивация, двигательные нарушения, резкое и прогрессирующее снижение когнитивных способностей. Болезнь продолжается от 8 до 72 месяцев (среднее значение составляет около 18 месяцев), после чего больной умирает. Происходит стремительная гибель нейронов и астроглиоз переднего и медиального таламуса и нижних оливок с последующим поражением коры головного мозга и мозжечка. Описана стадийность заболевания [18]: начальная форма характеризуется появлением тяжёлой бессонницы, панических атак, патологической тревоги и фобий; на второй стадии развития болезни больной начинает испытывать галлюцинации; на предпоследней стадии больной утрачивает способностью ко сну и стремительно теряет вес; терминальная стадия приводит к утрате речи и смерти.
В 2013 году была обнаружена иная клиническая форма прионных заболеваний, связанная с поражением вегетативной нервной системы [19]. Заболевание ассоциировано с появлением новой мутации в гене PRNP, приводящей к укорочению прионного белка и последующим нарушением его связи с клеточными мембранами. В этом случае распространение прионных агрегатов не ограничивается ЦНС. Предположительно, имеет место миграция PrPSc в переферические нервы и внутренние органы. Серия симптомов проявляется уже в раннем возрасте: хронический понос, вегетативная недостаточность и сенсорная полинейропатия. В зрелом возрасте наступает поражение ЦНС, приводящее к появлению деменции и судорог в числе симптомов.
Вариабельная протеаза-чувствительная прионопатия (VPSPr, ВПЧП) — ещё одно новое редкое спорадическое прионное заболевание, впервые описанное в 2008 году. VPSPr схожа с GSS по особенностям PrPSc, однако в прионе VPSPr не было детектировано каких-либо мутаций гена PRNP [20]. Невосприимчивость прионных белков, вызывающих VPSPr, к действию протеаз, значительно снижена. Болезнь манифестируется расстройствами речи (афазия, дизартрия), когнитивными нарушениями, в некоторых случаях атаксией и паркинсонизмом.
Перспективы лечения прионных заболеваний
Множество исследований в этой области дают основание для возможности ингибиции репликации прионов и лечения вызывамых ими болезней.
Как было отмечено выше, иммунный ответ организма на PrPSc отсутствует. Однако эксперимент, проведённый с полученными in vitro прионами, доказал, что использование антител, выработанных на определённые антигенные детерминанты PrP, индуцирует ингибирование размножения PrPSc [21], приводящее к отсрочке заболевания. Прионное превращение также может быть остановлено с помощью «блокаторов β-структур» — пептидных последовательностей, обогащенных аминокислотой пролином и обладающих гомологичным PrPC составом [5]. Иной подход основан на использовании антисенсовых олигонуклеотидов (АСО) — коротких фрагментов нуклеиновых кислот, останавливающих трансляцию с матричной РНК за счёт образования на ней петлеобразных участков. Именно АСО на данный момент является наиболее эффективным методом ингибиции репликации прионов: эксперименты с введением АСО в спинномозговую жидкость лабораторных мышей, проведённые в Лабораториях Скалистых Гор (Rocky Mountain Laboratories), привели к отсрочке проявления прионных болезней у подопытных на 113–135 дней [22]. Астемизол, относящийся к группе блокаторов H1-гистаминовых рецепторов (H1R), обладает доказанным антиприонным действием [23]. Кроме этого, для предотвращения размножения прионов возможно использование мутаций гена PNRP, приводящих к изменениям Q171R и E219K в аминокислотной последовательности PrP: мутантные прионные белки неспособны переходить в патологическую форму [24].
В данный момент лечение прионных заболеваний может быть только симптоматическим. Использование Брефельдина А, разрущающего аппарат Гольджи и тем самым замедляющего процесс распространения PrPSc и антагонистов NMDA-рецепторов, способствующих более длительному выживанию инфицированных клеток, в терапии CJD не достигло большого успеха [25]. Попытки применения классических противовирусных средств в лечении CJD и GSS также оказались неудачными [25]. Традиционные снотворные обладают нулевой эффективностью в терапии FSI и FFI, хотя был зафиксирован случай отсрочки летального исхода при одновременном использовании ряда сильнодействующих препаратов (диазепама, кетамина, оксида азота) [26]. Терапия VPSPr может быть основана на использовании повышенной чувствительности PrPSc к действию протеаз: доставка в организм смеси специфических иммобилизованных цистеиновых протеаз, вероятно, позволит отсрочить проявление болезни и пролонгировать жизнь больного. Этиотропное лечение основных ТГЭ, судя по всему, должно быть основано на использовании вышеперечисленных или подобных им методов ингибиции прионной репликации.
Основные термины (генерируются автоматически): CJD, PRNP, GSS, FFI, FSI, PNRP, белок, заболевание, болезнь, вегетативная нервная система.
Прионные болезни
В начале 1980-х годов в мировом научном мировоззрении произошел один из самых серьезных переворотов за всю историю биохимии, микробиологии, медицины, теории абиогенеза (возникновения жизни) и ряда других дисциплин. Однако человечество, к тому моменту уже практически утратившее способность удивляться, об этом открытии говорило недолго, – главным образом, в связи с эпизоотией «коровьего бешенства» 1986 года и жуткими документальными кадрами о заболевших и умирающих животных, – после чего все внимание отвлекли известные геополитические сдвиги того времени, открытый в тот же период ВИЧ и прочие события.
Между тем, работы исследовательской группы С.Прузинера (США, 1982-1984) не случайно были впоследствии удостоены Нобелевской премии. Речь шла ни много ни мало о том, что обнаружено «недостающее звено» между мертвой материей и органической жизнью, своеобразная переходная форма, – низкомолекулярное белковое соединение, способное размножаться за счет клетки-хозяина, не будучи при этом ни вирусом, ни бактерией, ни грибком, ни простейшим микроорганизмом. Более того, оказалось, что в точной репликации (воспроизведении) данного соединения не задействованы нуклеиновые кислоты, хотя до тех пор вненуклеиновая передача генетической информации для белковых форм жизни считалась в принципе невозможной.
История
Вероятно, часть болезней, которые еще в источниках IV-V веков упоминались под названиями «трясучая», «падучая» и т.п., имели прионную этиологию. Однако историю изучения заболеваний этой группы принято отсчитывать от начала ХVIII века, когда среди домашних животных разразился мор смертельной овечьей почесухи, или «скрейпи» (от англ. «to scrape», – скрестись, неистово чесаться, – что было одним из доминирующих симптомов), в Великобритании, затем во Франции, Германии («зудящая болезнь рысаков») и других западноевропейских странах.
В ХХ веке главными предпосылками прорывного открытия прионов можно считать первые клинические описания и исследования нейродегенеративного процесса, известного сегодня под названием болезнь Крейцфельдта-Якоба ; развитие концепции медленных вирусных нейроинфекций; изучение загадочно-эндемичной болезни куру в Папуа-Новой Гвинее и доказательство ее инфекционного характера (В.Зигас, К.Гайдузек, 1957, Нобелевская премия 1976 года); обнаружение сходных патоморфологических изменений у многих видов домашних и диких животных; работы британских микробиологов, которые с 1960-х годов предполагали наличие и пытались идентифицировать патоген, состоящий почти исключительно из белка, – некий инфекционный агент, очень устойчивый к физическим и химическим воздействиям (при которых погибают все прочие возбудители) и притом чрезвычайно малый, не достигающий даже размера вирусов. Завершили этот целенаправленный научный поиск, как указано выше, микробиологи из Калифорнийского университета в Сан-Франциско.
Этиопатогенез
Мембранно-клеточный белок PrP в организме млекопитающих распространен практически во всех тканях; у человека он кодируется особым геном PRNP в двадцатой хромосоме, причем биологическая роль и функции этого гликопротеина на сегодняшний день не вполне ясны.
Прион, или патогенный белок PrP Sc (можно встретить также обозначение PrP TSE ) – это структурно-искаженная, неправильно свернутая трансформация нормального белка PrP C . Данная структура, как и вирусы, не является клеткой; как и вироиды, не имеет оболочки; в отличие от любых других живых или условно-живых инфекционных агентов, не несет в себе рибонуклеиновый радикал. Указанные особенности исключают понимание прионов PrP Sc как чего-то живого. Единственная (по крайней мере, единственная известная на сегодняшний день) характеристика, роднящая прионы с другими формами жизни – это способность к самовоспроизведению: при контакте с «обычным» белком PrP прион каким-то образом превращает нормальные молекулы в точные копии собственных, деформированных, самовоспроизводящихся. Иными словами, это паразит, но паразит очень своеобразный, – не биологический, а биохимический, действующий на молекулярном уровне и смертельно опасный хотя бы в силу того, что иммунная система на него не реагирует. В результате пораженная ткань, – чаще всего это нейронные ткани центральной нервной системы, – лавинообразно вырождается с образованием микроскопических лакун и белковых отложений (см. также «Амилоидоз» , «Деменция с тельцами Леви» ). Задолго до открытия собственно прионов при патоморфологических исследованиях было установлено, что вещество мозга людей и животных, умерших от непонятных на тот момент болезней, имеет своеобразную микропористо-амилоидную структуру, – что и послужило основанием назвать прионные болезни спонгиоформными или спонгиозными (губчатыми, губкоподобными, губкообразными) энцефалопатиями. К настоящему времени в международном медицинском лексиконе устоялся термин трансмиссивная (лат. «передающаяся, инфекционная») спонгиоформная энцефалопатия, или ТСЭ (TSE).
Достоверно известно, что прионные заболевания могут наследоваться, возникать спонтанно или развиваться при проникновении прионов извне. Последний механизм, судя по всему, обусловил массовый падеж крупного рогатого скота от трансмиссивной губчатой энцефалопатии, и этим же путем (предположительно, при потреблении продуктов из содержащей прионы говядины) несколько человек заразились т.н. новым вариантом болезни Крейцфельдта-Якоба.
Конкретные причины деформации белковой молекулы, состав прионов, и, вообще, суть данного явления – все это остается предметом напряженных научных споров. Рассматривается несколько сильных гипотез, однако каждая из них сталкивается с не менее сильными контраргументами. В Международной классификации болезней десятого пересмотра (МКБ-10) прионные болезни были квалифицированы как «Атипичные вирусные инфекции центральной нервной системы» (все-таки вирусные), но в МКБ-11 это уже отдельная, отграниченная от вирусных инфекций рубрика.
Согласно имеющимся эпидемиологическим данным, – а они пока недостаточны, – чаще всего встречается спорадическая форма ТСЭ, обусловленная спонтанной мутацией гена PRNP. Заболеваемость в общей популяции оценивается на уровне примерно 1:1 000 000 новых случаев в год, при этом частота существенно варьирует в зависимости от региона (что тоже нуждается в исследованиях и объяснении). Наследственные формы являются более редкими, – как и случаи ТСЭ у человека, в отношении которых более-менее уверенно предполагается инфекционный характер, в том числе ятрогенный, связанный с введением различных зараженных прионами медицинских препаратов на основе животного биоматериала. Следует напомнить в этой связи, что прионы отличаются чрезвычайной стойкостью и не разлагаются при общепринятых, в том числе агрессивных видах обработки биологических образцов, а прионные болезни характеризуются атипично продолжительным инкубационным периодом (в некоторых случаях от заражения до клинической манифестации могут проходить десятки лет) в сочетании с быстрым катастрофическим протеканием (см. ниже).
Болезнь куру, которая столетиями была проклятием папуасского племени форе, оказалась именно трансмиссивной прионной инфекцией: обряды, практиковавшиеся этим племенем людоедов, подразумевали поедание головного мозга (в том числе детьми, проходящими ритуал инициации), в тканях которого и содержались патогенные формы прионного белка. К настоящему моменту это смертоносное эндемичное заболевание практически искоренено, – вместе с каннибализмом как таковым.
Однако о победе над прочими прионными болезнями говорить пока отнюдь не приходится. С большой долей вероятности перечень таких заболеваний (некоторым из них посвящены отдельные материалы на нашем сайте) будет расширяться; так, в прионную рубрику МКБ11 включено несколько различных вариантов болезни Крейцфельдта-Якоба , синдром Герстмана-Штросслера-Шейнкера, фатальная семейная бессонница , а также «другие», «уточненные» и «неуточненные» прионные заболевания человека.
Множество болезней, описанных и изучаемых современной медициной, остаются этиопатогенетически неясными. Часть из них – нейродегенеративные процессы, протекающие с отложением и накоплением амилоидных фибрилл, спонгиозом, глиозом (разрастание «каркасной» ткани нейронных структур) и другими патоморфологическими коррелятами, характерными и для прионных болезней. Поэтому правомерно и даже необходимо допустить, что прионный протеин PrP Sc – не единственная белковая молекула, способная размножаться в реакциях с гомологичными здоровыми молекулами. Представляется весьма вероятным, что для ряда «заболеваний неизвестной этиологии» в обозримом будущем будет обнаружен, описан и доказан механизм развития, аналогичный прионному. Так, в 2008 г. была открыта «вариабельная протеаза-чувствительная прионопатия», в 2013 г. официально признано существование «прионного заболевания, связанного с диареей и вегетативной нейропатией», и нет никаких оснований полагать, что список на этом закрыт.
Симптоматика
Клиническая картина прионных заболеваний различается в зависимости от преимущественной локализации и характера процесса (преобладание амилоидоза, глиоза и пр.), тогда как суть остается общей. Скажем, болезнь куру и очень медленно прогрессирующий синдром Герстмана-Штрауслера-Шейнкера в литературе рассматриваются как специфические разновидности болезни Крейцфельдта-Якоба.
Очевидно, однако, что описанное выше перерождение высокоорганизованных нервных тканей в любом случае должно сопровождаться развитием тяжелых психоневрологических нарушений и синдромов, – что и наблюдается в действительности. Спонгиоформная энцефалопатия проявляется прогрессирующими нарушениями праксиса (целенаправленные действия, движения), координации движений и пространственной ориентации (вплоть до тотальной астазии-абазии), зрительного восприятия (с развитием т.н. корковой слепоты), а также когнитивных функций, что в конечном счете приводит к тяжелой деменции (приобретенному слабоумию), полной беспомощности и несостоятельности больного. В некоторых случаях, например, при т.н. «новом варианте болезни Крейцфельдта-Якоба», заболевание манифестирует атипично рано, в молодом возрасте, и развивается на фоне длительно остающейся сохранной критики к собственному состоянию и перспективам. Однако в большинстве случаев ТСЭ диагностируют у лиц пожилого возраста, с некоторым (примерно в полтора раза) преобладанием мужчин.
В отсутствие этиопатогенетической терапии исход всегда летальный; на сегодняшний день спонгиоформные прионопатии, к сожалению, необратимы и неизлечимы. Продолжительность жизни после манифестации симптомов оценивается по-разному и зависит от формы заболевания, но в среднем составляет 1-3 года.
Диагностика
Диагностика прионных заболеваний остается серьезной проблемой для массовой клинической практики. Такие состояния встречаются редко, и мало кто из врачей имеет достаточный опыт в этом плане. Инкубационный период велик, что почти всегда исключает возможность найти причинно-следственную связь между каким-либо событием и началом патологического процесса (истинные причины вышеупомянутых эпизоотий специалисты пытаются установить до сих пор). Симптоматика неспецифична, те же клинические проявления могут встречаться при множестве других нейродегенеративных процессов. Пока не установлены достаточно надежные дифференциально-диагностические критерии.
И все же определенную настороженность у врача-невролога должны вызывать такие моменты, как быстрое прогрессирование когнитивных, моторных, перцептивных нарушений в отсутствие лабораторных признаков иммунной воспалительной реакции, а также определенная атипичность ЭЭГ. Для оценки состояния вещества мозга применяются визуализирующие методы (МРТ, ПЭТ); высокой информативностью и чувствительностью обладает анализ спинномозговой жидкости для исключения «обычных» инфекций и обнаружения белковых маркеров. Сообщается о разработке прогрессивных лабораторных методов, которые при широком внедрении позволят выявлять патогенные прионы на самых ранних доклинических стадиях и с практически стопроцентной надежностью.
Однако сегодня во многих случаях спонгиоформная энцефалопатия достоверно диагностируется лишь в ходе посмертной аутопсии.
Лечение
Как указано выше, этиопатогенетической терапии в настоящее время нет; остановить размножение прионных молекул или, тем более, обратить вспять злокачественную деструкцию нейронных тканей существующими сегодня способами невозможно. Применяются те или иные схемы поддерживающей, паллиативной, симптоматической терапии с использованием нейропротекторов, стимуляторов мозговой трофики и некоторых других препаратов, в отношении которых получены предварительные данные об их антиприонном эффекте. Работы в данном направлении ведутся сейчас крупнейшими научно-исследовательскими центрами и фармацевтическими гигантами, поскольку, – подчеркнем это в заключение, – заболевания прионного типа могут оказаться гораздо более распространенной проблемой, чем представлялось еще 20-30 лет назад, и медицина должна быть к этому готова.
Прионы – особый класс возбудителей медленных инфекций человека и животных
Для цитирования: Зуев В.А. Прионы – особый класс возбудителей медленных инфекций человека и животных. РМЖ. 2013;30:1559.
Формальное знакомство с прионными болезнями уходит своими корнями в далекое прошлое, когда более 280 лет назад была описана в Англии (1732 г.), а вскоре и в Германии (1750 г.) одна из «классических» прионных болезней – скрепи у овец. Эта болезнь с 1755 г. уже столь широко распространена, что становится даже предметом петиции в Британский парламент, поданной фермерами-овцеводами графства Линкольншир. Только в 1899 г. удается доказать инфекционную природу скрепи. Постепенно болезнь широко распространяется среди племенных овец и в различных странах получает свои названия [1].
Литература
1. Тимаков В.Д., Зуев В.А. Медленные инфекции. М.: Медицина, 1977. С. 7–17.
2. Sigurdsson B.// Brit. Vet. J. 1954. Vol. 110. P. 255–270.
3. Idem ibid. P. 307–322.
4. Idem ibid. P. 341–354.
5. Gajdusek D.C., Gibbs C.J. Slow, latent, and temperate virus infections; eds. D.C. Gajdusek, C.J. Gibbs, M. Alpers. Monograph. № 2. Washington, 1965. P. IX–X.
6. Sigurdsson B., Thormar H., Polson P.A. // Arch. ges. Vidusforsch. 1960. Vol. 10. P. 368–381.
7. Horta-Barbosa L., Fucillo D., Sever J. et al. // Nature. 1969. Vol. 221. P. 974.
8. Weller T.H., Neva F.A. // Proc. Soc. Exp. Biol. Med. 1962. Vol. 3. P. 215–225.
9. Зуев В.А. Медленные вирусные инфекции человека и животных. М.: Медицина, 1988. C. 115–136.
10. Сuille J., Chelle P.L. C.R. // Acad. Sci. 1936. Vol. 203. P. 1552–1554.
11. Gajdusek D.C., Zigas V. // New Engl. J. Med. 1957. Vol. 257. P. 974–978.
12. Alpers M.P. // Am.J. trop. Med. Hyg. 1970. Vol. 19. P. 133–137.
13. Lampert P.W., Gajdusek D.C., Gibbs C.J. // Am. J. Pathol. 1972. Vol. 68. P. 626–646.
14. Hadlow W.J. // Lancet. 1959. Vol. 2. P. 289–290.
15. Gibbs C.J., Gajdusek D.C., Asher D.T. et al. // Science. 1968. Vol. 161. P. 388–389.
16. Alperovich A. // Eur. J. Neurol. 1996. Vol. 3. P. 500–506.
17. Brown P., Gibbs C.J., Rodgers-Johnson P. et al. // Ann. Neurol. 1994. Vol. 35. P. 513–529.
18. Duffy P., Wolf J., Collins G. et al. // New Engl. J. Med. 1974. Vol. 290. P. 692–693.
19. Bradley R. Prion Diseases; td. J. Collinge and M.S. Palmer. Oxford Univ. Press. 1997. P. 89–129.
20. Williams E.S., Young S. J. // Wildi Dis. 1980. Vol. 16. P. 89–98.
21. Gajdusek D.C. Virology; ed. B.N. Fiedds. New York,1985. P. 1519–1557.
22. Gajdusek D.C. Subviral Parthogenesis of Plants and Animals: Viroid and Prions. New York, 1985. P. 483–544.
23. Prusiner S.B., McKinley M.P., Groth D.F. et al. // Proc. Natl. Acad. Sci. 1981. Vol. 78. P. 6675–6679.
24. Prusiner S.B. // Science. 1982. Vol. 216. P. 136–144.
25. Bolton D.C., McKinley M.P., Prusiner S.B. // Scvience. 1982. Vol. 218. P. 1309–1311.
26. McKinley M.P., Bolton D.C., Prusiner S.B. // Cell. 1983. Vol. 35. P. 57–62.
27. Imran M., Mahmood S.V. // Virol. Journal. 2011. Vol. 8. P. 559.
28. Tobler J., Gaus T., Deboer P. et al. // Nature. 1996. Vol. 380. P. 639–642.
29. Harris D.A., Falls D.L., Johnson F.A. et al. // Proc. Natl. Acad. Sci. 1991. Vol. 88. P. 7664–7668.
30. Lugaresi E., Medori P., Baruzzi A. et al. // New Engl. J. Med. 1986. Vol. 315. P. 997–1003.
31. Boldin E., Capellari C., Provini F. et al. // J. Neurol. 2009. Vol. 256. P. 778–779.
32. Зуев В.А., Завалишин И.А., Ройхель В.М. Прионные болезни человека и животных. М.: Медицина, 1999. C. 136–142.
33. Bradley R. Prion Diseases; eds. J. Collinge, M.S. Palmer. Oxford, 1997. P. 89–129.
34. Покровский В.И., Киселев, Черкасский Б.Л. Прионы и прионные болезни. М.: Изд-во РАМН, 2004. C. 45–55.
35. Colby D.W., Prusiner S.B. Prions. // Cold Spering Harb. Perspect. Biol. 20011. Vol. 3. P. 1–22.
36. Chazot G., Brousolle E., Lapras C.I. et al. // Lancet. 1996. Vol. 347. P. 1181.
37. Collinge J., Palmer M. Prion Diseases; eds. J. Collinge, M. Palmer. Oxford, 1997. P. 18–55.
38. Parchi P., Saverioni D. // Folia Neuropathol. 2012. Vol. 50(1). P. 20–45.
39. Григорьев В.Б., Покидышев А.Н., Кальков С.Л. и др. // Вопр. вирусол. 2009. T. 5. P. 4–9.
40. Трубачева Е.С. Материалы 2-й Межрегиональной научно-практич. конф. «Тольяттинская осень». Тольятти, 2009. C. 296–299.
41. Brown H. // Antiviral. Chem. Chemother. 1990. Vol. 1. P. 75–83.
42. Gambetti P., Cali I., Notari S. et al. // Acta Neuropathol. 2011. Vol. 121 (1). P. 79–90.
43. Safar J.G. // Prion. 2012. Vol. 6(2). P. 108–115.
44. Зуев В.А., Игнатова Н.Г., Автандилов Г.Г. // Успехи геронтол. 2005. T. 17. C. 108–116.
45. Villeda S., Luo J., Mosher K. et al.// Natura. 2011. Vol. 477. P. 90–96.
46. Carp R.A. Int. Virol. 2nd int. // Congr. Virol. Budapesht. 1971. Karger. Basel. 1972. P. 210.
47. Haralambiev H., Ivanov I., Vesselinova A. // Zbl. Vet. 1973. Vol. 201. P. 201–209.
48. Ройхель В.М., Фокина Г. И., Кондакова Л.И. // Вопр. вирусол. 1997. № 5. C. 203–205.
49. Gambetti P., Dong Z., Yuan J. et al. // Annal. Neurol. 2008. Vol. 63. P. 697–708.
50. Zou W.Q., Puoti G., Xiao X. et al. // Annal. Neurol. 2010. Vol. 68. P. 162–172.
51. Bendheim P.E., Barry R.A., DeArmond S.J. et al. Antibodies to a scrapie prion protein // Nature. 1984. Vol. 310(5976). P. 418–421.
52. Ройхель В.М. Медленные болезни человека и животных, вызванные прионами // Природа. 2002. № 2.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Правда ли, что через вакцину от коронавируса можно заразиться прионными заболеваниями?
Первоисточником информации о грядущих побочных явлениях вакцинации стали люди с медицинским и биологическим образованием. Американский иммунолог Джон Классен предупреждает о таких последствиях мРНК-вакцин в своей статье «РНК-вакцины против COVID-19 и риск прионных заболеваний», которую опубликовал журнал Microbiology & Infectious Diseases. Российский микробиолог и полковник медицинской службы запаса Михаил Супотницкий сообщает в своей книге «COVID-19: трудный экзамен для человечества», что к таким последствиям может привести S-белок из вакцин. Французский нобелевский лауреат Люк Монтанье заявляет, что знает о как минимум пяти случаях прионного заболевания, возникшего после вакцинации препаратом Pfizer — BioNTech.
Информацию о связи вакцин и прионных заболеваний также распространяют в Telegram-каналах, блогах, социальных сетях и даже в СМИ. Например, актриса и телеведущая Мария Шукшина утверждает: «Экспериментально показано, что S-белок обладает прионным эффектом, то есть вызывает неправильную укладку белков нейрональных клеток и повреждает нейроны мозга. В крови он индуцирует образование нерастворимых амилоидных сгустков, мешающих кровотоку и вызывающих тромбозы сосудов. В отдалённой перспективе — развитие прионных болезней мозга у вакцинированных. Игнорирование этого свойства S-белка приведёт к тому, что через несколько лет у десятков миллионов вакцинированных россиян будет выявлено слабоумие и нейродегенеративные заболевания мозга». Об РНК-вакцине сообщают, что она «вызывает долгосрочные неврологические нарушения вплоть до смертельных болезней Альцгеймера и Лу Герига». По словам пользователей интернета, у некоторых вакцинированных слабоумие наступало буквально через два дня после прививки.
Прионные болезни — группа нейродегенеративных заболеваний человека и животных, вызываемых прионами, особыми инфекционными белками.
Первым описанным прионным заболеванием была болезнь овец под названием «скрепи», выражающаяся вначале кожным зудом, из-за чего животное начинало чесаться (scrape — англ. «скрести»), затем наступал паралич и смерть. Случаи скрепи отмечались ещё в 1700-е годы в Европе. В 1920-х немецкие врачи Ганс-Герхард Крейтцфельдт и Альфонс Якоб исследовали неизлечимое заболевание нервной системы у людей. В частности, они сделали препараты мозга погибших, похожие на губку из-за гибели целых участков нейронов. Позже заболевание было названо болезнью Крейтцфельдта — Якоба в честь её исследователей.
Только в 1967 году американский педиатр Карлтон Гайдушек смог связать клиническую картину другого прионного заболевания, болезни куру, и возбудителя, не относящегося к ранее известным бактериями, вирусам, простейшим или грибкам. Гайдушек изучал в Новой Гвинее вирусные заболевания. Там он заинтересовался болезнью, которые аборигены называли куру — на местном языке это слово обозначало «дрожь» или «порчу», что указывало на веру в сверхъестественную природу заболевания. Наиболее подвержены куру были дети и женщины племени форе, практиковавшего ритуальный каннибализм. На протяжении нескольких месяцев заболевшего человека трясло, из-за дрожи он терял способность вначале стоять, потом и сидеть, затем дрожь сменял паралич и наступала смерть.
Препараты мозга форе, отправленные в Европу, были крайне похожи на те, что заготовили в своё время Крейтцфельдт и Якоб. Их структуру также опознал британский ветеринар Билл Хэдлоу, указав, что наблюдал подобные поражения у овец, погибших от скрепи. Так все эти заболевания были объединены в одну группу. Карлтон Гайдушек и работавшая там же антрополог Ширли Гласс догадались, что ритуальный каннибализм и есть путь передачи заболевания. Из уважения к погибшему соплеменники съедали его, в том числе мозг, так как верили, что его таланты таким образом передаются другим. Женщины и дети, заболевавшие чаще всего, как раз занимались приготовлением ритуальной поминальной трапезы, а следовательно, и больше контактировали с инфицированными тканями. На сегодняшний день куру в племени форе удалось искоренить почти полностью, избавившись от ритуального каннибализма. По разным данным, последняя смерть от куру зафиксирована в 2005 или 2009 году.
Однако возбудитель болезни долгое время оставался неизвестным — на него не действовали известные антибиотики, он не погибал при температуре 95 °С и не распознавался под микроскопами того времени. Тогда Карлтон Гайдушек и предположил наличие некого патогена, меньше вируса по размеру. Только в 1982 году частицу смог выделить американский врач Стэнли Прузинер, он назвал её прионом (от слов protein, «белок», и infection, «инфекция»). За это открытие он был удостоен Нобелевской премии по физиологии и медицине в 1997 году.
На сегодняшний день известно пять заболеваний человека, которые вызывают прионы: уже упомянутые выше болезнь Крейтцфельдта — Якоба и куру, а также синдром Гертсманна — Штреусслера — Шейнкера, вариабельная протеаза — чувствительная прионопатия и фатальная инсомния (все три имеют преимущественно наследственную природу, и лечения от них не существует). У животных прионы ответственны за возникновение трансмиссивных губчатых энцефалопатий, в том числе губчатой энцефалопатии крупного рогатого скота (известной в народе как коровье бешенство). Сейчас прионные заболевания обладают 100-процентной летальностью, при этом от заражения до появления симптомов обычно проходит около 30 лет, есть, правда, и более быстро развивающиеся формы.
Прион — белок, производство которого закодировано в 20-й хромосоме, чья нормальная форма может спонтанно или под действием внешних факторов менять свой химический состав, вызывая нейродегенеративные заболевания. Болезни Альцгеймера и Лу Герига (также известна как боковой амиотрофический склероз, БАС) не относятся к прионным заболеваниям. Основной причиной первой на сегодняшний день считают отложения бета-амилоида, пептида, чей белок-предшественник закодирован не в 20-й, а в 21-й хромосоме. Причина БАС окончательно не установлена, около 20% случаев — наследственные, связанные с мутациями гена супероксиддисмутазы-1, расположенного опять же не в 20-й, а в 21-й хромосоме.
Так как авторы заявлений говорили о разных вакцинах и смешали вместе прионные и другие нейродегенеративные заболевания, мы рассмотрим отдельно четыре утверждения:
— вакцины на основе S-белка вызывают прионные заболевания;
— вакцины на основе S-белка вызывают нейродегенеративные заболевания, в частности болезни Альцгеймера и Лу Герига;
— вакцины на основе мРНК вызывают прионные заболевания;
— вакцины на основе мРНК вызывают нейродегенеративные заболевания, в частности болезни Альцгеймера и Лу Герига;
К вакцинам на основе S-белка относится используемый в России «Спутник V», а также некоторые западные, например AstraZeneca и Johnson & Johnson. S-белок по своей структуре даже не близок к белку PrPC (нормальный прионный белок) или к белку PrPSc (прионный белок, связанный с заболеваниями). Самое наглядное их различие: S-белок состоит из порядка 1300 аминокислотных оснований, а PrPC — лишь из 208. Обладать прионным эффектом неприонный белок не может.
Самый подробный отчёт по побочным явлениям от вакцин, включающий в себя данные о препаратах, основанных на S-белке, выпущен министерством здравоохранения Аргентины. Всего в нём проанализированы возможные побочные действия после более 13 млн доз «Спутника V» и 14 млн AstraZeneca. При этом ни одна из этих вакцин, согласно отчёту, не стала причиной развития болезней Альцгеймера или Лу Герига. Сразу стоит отметить, что упоминаний прионных заболеваниях там тоже нет. Таким образом, никакие научные данные не подтверждают теории, изложенные Михаилом Супотницким .
Вакцины на основе мРНК в России не используются. Этот класс вакцин представлен двумя препаратами: американской Moderna и немецко-американской Pfizer — BioNTech. В американской системе регистрации побочных явлений VAERS нет информации ни о прионных, ни о нейродегенеративных заболеваниях, возникших из-за вакцинации мРНК-вакцинами, сообщила американскому фактчекинговому ресурсу PolitiFact Марта Шаран, представитель Центров по контролю и профилактике заболеваний США (CDC). Американская ассоциация по болезни Альцгеймера настоятельно рекомендует вакцинироваться пациентам, имеющим это заболевание.
Прионное заболевание, в частности болезнь Крейтцфельдта — Якоба, имеет четыре причины развития: спорадическая (ген мутирует сам по себе) — около 85% случаев; наследственная (ген изначально более склонен к мутации) — по разным оценкам, от 5% до 15%; ятрогенная (заражение мутировавшим прионом происходит извне, например при каннибализме у форе или при некачественно проведённой медицинской процедуре) — менее 1%; новая, вариантная (при употреблении в пищу мяса инфицированного скота) — известно 178 случая в Великобритании и 53 за её пределами. Логично, что хоть как-то с вакцинацией может быть связана только ятрогенная форма. Однако это опровергается несколькими фактами.
Ещё Карлтон Гайдушек потерпел неудачу, пытаясь заразить животных с помощью инъекций частиц крови и мышц и даже препаратов мозга от погибших людей. Первый удачный запрограммированный случай заражения произошёл, лишь когда он ввёл кашицу из мозжечка погибшего от куру прямо в мозг подопытной шимпанзе Жоржетте. При этом клетки мозга не входят в состав тех клеточных линий, которые действительно использовались во время разработки почти всех известных на сегодняшний день вакцин против коронавируса.
Более того, большинство готовых препаратов (в частности, используемая в России основанная на S-белке вакцина «Спутник V», неиспользуемая в России основанная на S-белке AstraZeneca, а также основанные на мРНК-технологии Moderna и Pfizer — BioNTech) очищены от клеточной линии хроматографическим методом. Более того, использованные при разработке этих вакцин линии, преимущественно HEK (Human Embryonic Kidney) 293, принадлежат, как видно из названия, эмбрионам (врождённая форма болезни Крейтцфельдта — Якоба науке не известна), следовательно, они не могут быть инфицированы прионом. К тому же линией HEK 293 пользуются с 1973 года, то есть уже 48 лет, и ни одного случая развития болезни из-за вакцинации не зафиксировано.
При этом в истории человечества были случаи заражения прионными болезнями в ходе медицинских процедур. Например, во Франции в результате заражения в 1980-е погибло 110 детей (отстающим в росте детям вводили препарат из гипофиза животных, больных коровьим бешенством), и четыре случая заражения случились из-за переливания крови между 1996 и 1999 годом. После этого нет данных ни об одном случае ятрогенной передачи болезни. Отдельно стоит отметить, что авторитетным американским медицинским ресурсам и до начала пандемии новой коронавирусной инфекции приходилось развеивать опасения граждан насчёт того, может ли вакцина стать источником коровьих прионов. Ответ однозначен: нет, не может. При производстве вакцины действительно может использоваться сыворотка, получаемая из тканей коров. Однако прионы имеются только в мозге, спинном и головном, и в сетчатке. В сыворотке же используется эмбриональная кровь. Мало того что прионы вообще не содержатся в крови, плацентарный барьер также обеспечивает фильтрующую функцию, делая невозможным передачу прионов от инфицированного животного нерожденному эмбриону.
Кожа и соединительная ткань, которые используются для приготовления желатина, на котором выращивают клеточную культуру, также не может содержать прионов. Отдельно учёные обращают внимание, что «передача прионов ни разу не была задокументирована после внутримышечной или подкожной инъекции вакцины». CDC ещё в 2000 году выпустили документ, в котором сообщается, что «нет ни одного доказательства о связи болезни Крейтцфельдта — Якоба и вакцинации». Риск заболеть после прививки они назвали лишь «теоретическим», подчеркнув, что преимущества вакцинации его значительно перевешивают.
Отдельно стоит рассмотреть, где и кто опубликовал эту ложную информацию о связи вакцин с прионными и нейродегенеративными заболеваниями. История об этой связи относительно мРНК-вакцин была опубликована Джоном Классеном в журнале Microbiology & Infectious Diseases, который относится к так называемым «хищническим журналам» из списка Джеффри Билла. За публикации у себя они берут с авторов деньги, при этом не обеспечивают необходимого редактирования и научного рецензирования. То есть информации, опубликованной в таком журнале, доверять в принципе нельзя. Отдельно стоит отметить, что импакт-фактор этого журнала составляет лишь 0,29.
У научного сообщества есть претензии и к тексту статьи. Так, автор пишет, что мРНК-вакцина «была проанализирована на содержание последовательности, которые индуцируют сворачивание TDP-43 и FUS в их патологические прионные структуры, что приводит к развитию БАС, дегенерации передней височной доли, болезни Альцгеймера и другим неврологическим заболеваниям». При этом в самом исследовании нет никаких данных, демонстрирующих, что за анализ был проведён. Учёный и популяризатор науки Дэвид Горски критикует работу Джона Классена: «Серьёзно, как он сделал это? Он не говорит. Он просто утверждает, что "проанализировал" последовательность мРНК, кодирующую спайковый белок SARS-CoV-2, который использовался в вакцине Pfizer — BioNTech COVID-19, но не объясняет, как он это сделал. Он не приводит никаких данных, демонстрирующих, как эта последовательность активирует TDP-43 и FUS». Американский совет по вопросам науки и здравоохранения также выпустил статью-опровержение, в которой разобрал основные тезисы автора материала.
Если посмотреть внимательно на биографию Классена, то в ней можно найти его и более ранние антипрививочные заявления. В частности, в 1999 году он утверждал, что есть связь между вакцинацией от гриппа и диабетом I типа. При этом в Стандартах медицинской помощи при диабете, выпущенных Американской диабетической ассоциацией, подчёркивается важность иммунизации этой группы населения против гриппа. Институт по безопасности вакцин Университета Джонса Хопкинса сделал даже систематический обзор, включающий 13 исследований, который демонстрирует, что никакой связи между прививкой от гриппа и диабетом нет. Помимо этого, Классен активно распространяет информацию об опровергнутой наукой связи между вакцинацией и ожирением, а также развитием аутизма у детей. Идеи Классена активно продвигает антипрививочник и спонсор этого движения Роберт Кеннеди-младший, подробнее о деятельности которого и заработках на этой теме можно прочитать у наших коллег.
Люк Монтанье, хоть и получил Нобелевскую премию по физиологии и медицине в 2008 году, во время пандемии новой коронавирусной инфекции был не раз пойман фактчекерами на распространении дезинформации. Свои слова о пяти людях, заражённых болезнью Крейтцфельдта — Якоба, он ничем не подтверждает. В отчёте CDC о побочных явлениях нет подобных данных. Сама картина заболевания (клинические симптомы развиваются через много лет после инфицирования) противоречат словам нобелевского лауреата.
Михаил Супотницкий не имеет такого научного признания, как Люк Монтанье, и не публиковал свою теорию в научных журналах. Он выпустил свою книгу в издательстве «Русская панорама», которое, согласно заявлению на сайте, сотрудничает с «вузами и академическими институтами». Несмотря на звание полковника медицинской службы и должность заместителя главного редактора одного из научных журналов Министерства обороны РФ, он придерживается не признаваемых научным сообществом взглядов на эпидемию ВИЧ/СПИД. В своей книге он также придерживается не нашедшей научного подтверждения точки зрения об антителозависимом усилении.
Подводя итоги: болезни Альцгеймера и Лу Герига не относятся к прионным заболеваниям вообще. Вакцины, основанные на S-белке или на мРНК, не способны вызвать ни их, ни настоящие прионные заболевания. Распространяющие эти истории учёные не раз уже замечены фактчекерами в конспирологии, их работы публикуются в неавторитетных изданиях, а высказанные ими гипотезы не находят подтверждения у научного сообщества.
Читайте также: