Влияние гипофосфатемии на сердце. Воздействие ацетальдегида на сердце
Добавил пользователь Владимир З. Обновлено: 30.01.2026
Латентная стадия алкогольной кардиомиопатии. Атеросклероз как причина алкогольной кардиомиопатии
Эхокардиографическое обследование 13 добровольцев показало, что нагрузка этанолом вызывает угнетение сократительной функции миокарда, проявляющееся наиболее отчетливо уменьшением фракции выброса (отношения ударного объема к конечному диастолическому объему) и средней скорости кругового укорочения волокон миокарда; эти показатели отклонялись от данных контроля в течение 2 ч острой интоксикации и возвращались к норме на 150-й минуте опыта.
Исследования, проведенные у больных с хронической алкогольной интоксикацией, позволили сделать вывод, что нарушения сократительной функции миокарда начинают определяться до появления клинических признаков сердечной недостаточности и поражения сердца вообще, что свидетельствует о наличии доклинической, или латентной (А. М. Скупник, 1974), стадии алкогольной кардиомиопатии.
Изучение кардиодинамики с помощью катетеризации сердца подтверждает результаты неинвазивных методов исследования: алкоголь в малых дозах у здоровых людей вначале может несколько увеличить показатели работы левого желудочка, по-видимому, за счет гиперкатехоламинемии (М. Zahir с соавт., 1971), но большие дозы, а также алкогольная нагрузка у больных с неалкогольными заболеваниями сердца, с алкогольной кардиомиопатией, у больных с тяжелым алкогольным циррозом печени неизменно приводят к снижению сердечного индекса и ударного объема и повышению конечного диастолического давления в левом желудочке.
Хотя к кардиомиопатиям принято относить заболевания мышцы сердца, протекающие без атеросклероза коронарных артерий, существует мнение, что кардиомиопатия является следствием стенозирующего поражения мелких мышечных веточек венечных артерий (А. В. Виноградов, Г. А. Глезер и В. С. Жданов, 1973). Коронарные артерии при алкогольной кардиомиопатии по данным коронарографии не поражаются (R. Haasis, D. Larbig и D. Jeschke, 1976).
Однако хорошая проходимость крупных коронарных артерий, проявляющаяся «нормальной» коронарной ангиограммой, еще не исключает возможности поражения мелких артерий (G. Burch и Т. Giles, 1977). J. Goodwin (1973) и S. Factor (1976) указывают, что неспецифические изменения миокарда при алкогольной кардиомиопатии могут быть результатом вторичной ишемии миокарда вследствие поражения небольших интрамуральных разветвлений коронарных артерий и артериол. В этих сосудах S. Factor обнаружил отек стенки, периваскулярный фиброз, участки гиалиноза, преимущественное поражение эндотелия, сопровождающееся повышением проницаемости сосудистой стенки.
Подобные морфологические изменения, в особенности повышение сосудистой проницаемости при алкогольной интоксикации, были еще раньше описаны О. Б. Мазиковой (1954, 1958), В. И. Яковлевой (1967), Г. П. Казанцевой (1975, 1976) и другими отечественными исследователями. Однако пока еще нет достаточных оснований считать, что главную патогенетическую роль в формировании алкогольной кардиомиопатии играют поражения мелких коронарных артерий. Тщательно изучив состояние малых артерий миокарда у 158 больных, умерших от различных заболеваний, в том числе в двух случаях от алкогольной кардиомиопатии, J. Burch и Т. Giles (1977) констатировали нормальную пространственную архитектуру малых артерий, пенетрирующих в глубину желудков и межжелудочковой перегородки при алкогольном сердце, и лишь отметили, что правый желудочек был васкуляризирован хуже левого.
Возможно, большую роль в патогенезе поражения сердца при алкогольной интоксикации играют нарушения микроциркуляции (повышение проницаемости капилляров и перикапиллярный отек, снижение кровотока, местами агрегация эритроцитов), причем нарушение капиллярного кровотока обнаруживается в доклинической стадии болезни. С помощью электрофизиологических исследований в эксперименте и в клинике обнаружено, что алкоголь влияет не только на сократительную функцию миокарда, но и на проводящую систему сердца.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Влияние гипофосфатемии на сердце. Воздействие ацетальдегида на сердце
Несомненную роль в нарушении механизмов сокращения мышечных волокон может играть и свойственная хронической алкогольной интоксикации гипофосфатемия, ведущая к повреждению синтеза АТФ и 2,3-дифосфоглицерата (N. Buchbinder и W. Roberts, 1974).
Острая алкогольная интоксикация возбуждает симпатико-адреналовую систему, вызывая быстрое увеличение уровня катехоламинов в крови. Одновременно повышается экскреция гликокортикоидов (A. Anton, 1951; G. Santisteban и С. Swinyard, 1955; R. Maickel и R. Paoletti, 1961), которые сенсибилизируют миокард к деструктивному действию катехоламинов (W. Raab, 1966; Т. lames и Е. Bear, 1967). Хроническая алкогольная интоксикация сопровождается гипокортицизмом и истощением миокардиальных и надпочечниковых запасов катехоламинов (J. Wartburg, W. Berli), что приводит к снижению инотропного влияния симпатического отдела нервной системы на гипертрофированный миокард, снижает его адаптационные возможности и является еще одной причиной недостаточности сердца.
Однако большинство исследователей основное значение в продуцировании патологических изменений миокарда придают непосредственному действию алкоголя и его первого метаболита — ацетальдегида — на мышцу сердца. Это подтверждается многочисленными исследованиями как экспериментаторов, так и клиницистов с использованием многочисленных современных методик. В 1946 г. К. Wakim, перфузируя раствор этилового алкоголя через изолированное сердце черепахи, получил вначале снижение амплитуды сердечных сокращений, а затем, по мере увеличения концентрации раствора, дилатацию сердца и остановку его в диастоле; часто при этом развивалась фибрилляция желудочков.
Y. Hirota, О. Bing и W. Abelman (1976) помещали трабекулы и папиллярные мышцы сердца крысы в оксигенируемый раствор Кребса и регистрировали параметры сокращения и последующего расслабления ткани при искусственной стимуляции. Добавление этанола вызывало уменьшение максимального напряжения соответственно концентрации алкоголя. W. Newman и J. Valicenti (1971) в опыте на собаках демонстрировали ослабление систолы левого желудочка, которая снова усиливалась при введении вслед за алкоголем уобаина. Т. Regan с соавторами (1974) в эксперименте на собаках, получавших алкоголь при адекватном питании в течение двух лет, показали выраженное нарастание конечного диастолического давления в левом желудочке и значительное снижение ударного объема сердца при нагрузке ангиотензином в сравнении с контрольными животными.
Для исключения экстракардиальных явлений, сопровождающих острую алкогольную интоксикацию, в особенности действуя на сердце катехоламинов при алкогольной нагрузке, М. Wong (1973), L. Horwitz и J. Atkins (1974) проводили свои эксперименты на собаках, которым перед введением алкоголя производилась комбинированная бета-адренергическая и постганглионарная парасимпатическая блокада пропроналолом и атропина сульфатом. В связи с тем что депрессия сердца под влиянием этанола была демаскирована автономной блокадой, нарастание конечного диастолического давления и увеличение диастолического и систолического внутреннего диаметра левого желудочка при снижении сердечного индекса и ударного объема сердца были выражены особенно отчетливо.
Изменение механизма сокращения сердца под влиянием алкоголя у здоровых и у страдающих хроническим алкоголизмом изучалось в основном неинвазивными методами — с помощью поликардиографии, механокардиографии, определения величины сердечного выброса методом разведения красителя,, эхокардиографии. При поликардиографических и баллистокардиографических исследованиях отмечалось увеличение фазы напряжения и укорочение времени изгнания как у здоровых, так и у больных хроническим алкоголизмом, а также у лиц с заболеваниями сердца неалкогольной этиологии при введении им этилового спирта.
Баллистокардиографическая кривая в ранних стадиях алкогольного поражения сердца часто была высокоамплитудной, гиперкинетической, отражая гиперкинетический тип циркуляции, в поздних стадиях болезни — низковольтной, гипокинетической, часто хаотической. Сердечный выброс снижался пропорционально тяжести поражения миокарда.
Клиника алкогольной кардиомиопатии. Признаки поражения сердца алкоголем
Впервые подробно описал клинику алкогольного поражения миокарда G. Steel (1893). J. Mackenzie (1902) в своем описании клиники алкогольной кардиомиопатии, не устаревшем, по мнению Е. М. Тареева, до настоящего времени, обращал внимание на основные симптомы болезни — учащение пульса и одышку при усилии: «Во всех непонятных случаях учащенной деятельности сердца следует тщательно выделить вопрос относительно злоупотребления алкоголем.
Больные часто стараются скрыть свои привычки в этом отношении, но врачу обыкновенно удается найти ключ к решению в манере больного держать себя, в выражении его лица, дрожании мышц (особенно языка), отсутствии аппетита или тошноте по утрам и во всем том ансамбле, по которому опытный врач заподозривает тайного алкоголика. Учащенный пульс и другие явления часто сопровождаются ощущением замирания под ложечкой и чувством изнеможения и одышки при усилиях. Сердце может быть немного увеличено или же бывает сильно расширено, что обыкновенно сопровождается увеличением печени и чувствительностью покрывающих ее тканей.
При воздержании от алкоголя такие больные, в ранних стадиях, быстро поправляются, но с продолжением алкоголизма развиваются все характерные черты тяжелой недостаточности сердца».
Чрезвычайно ценным является также указание J. Mackenzie на то, что декомпенсация может некоторое время протекать в скрытой форме, но быстро выявляется при приступах пароксизмальной тахикардии, когда серДце, почти нормальное по размерам, в течение нескольких часов значительно увеличивается, лицо больного становится синюшным, губы распухают, шейные вены сильно пульсируют.
В течение суток появляется отек ног, увеличивается печень. С прекращением приступа пароксизмальной тахикардии больные тотчас же испытывают облегчение и через несколько часов все следы недостаточности сердца исчезают, а сердце приобретает прежние размеры и нормальный ритм.
В 1921 г. Н. Vaquez описал подострый алкогольный миокардит, который долгое время может оставаться нераспознанным ввиду медленного развития и начинается постепенно прогрессирующей одышкой, сердцебиением, отеком ног, болезненной застойной печенью, легкой желтушностью, сосудистой гипертонией, неритмичным пульсом, ритмом галопа или систолическим шумом функциональной недостаточности митрального клапана.
В этот период болезнь еще можно остановить, хотя выздоровление будет неполным (пульс чаще остается неправильным и учащенным). Если же больной продолжает злоупотреблять алкоголем, недостаточность сердца прогрессирует, к шуму митральной недостаточности присоединяется шум недостаточности трикуспидальной; в легких появляются хрипы и очаги инфарктов с кровянистой мокротой; больные погибают от сердечного коллапса или внезапно во время обморока.
Механизм патогенного влияния алкоголя рассматривался в тот период с точки зрения как непосредственного действия его на миокард, так и гемодинамической перегрузки сердца большим количеством жидкости (вина, пива).
Проблема алкогольной кардиомиопатии
В настоящее время, в связи с широчайшей распространенностью алкоголизма в России, проблема своевременной диагностики и лечения заболеваний, ассоциированных с чрезмерным приемом спиртных напитков, стоит особо остро. Темпы прироста смертности, связанной с употреблением алкоголя в нашей стране, особенно в выходные дни, критически высоки, и мировое медицинское сообщество выражает озабоченность по этому поводу (12,22,24).
Несмотря на то, что алкогольная кардиомиопатия (АКМП) четко описана и с 1996 г. рассматривается ВОЗ в рамках дилатационной кардиомиопатии, а согласно МКБ выделена в отдельную нозологическую форму (I42.6), существуют определенные трудности в практической постановке данного диагноза, как в клинике, так и на секционном столе. Вразумительные схемы лечения АКМП на сегодняшний день не разработаны. Целью данного обзора является попытка рассмотреть с клинической точки зрения сущность феномена развития кардиомиопатии на фоне злоупотребления алкоголем.
МЕСТО АЛКОГОЛЬНОЙ КАРДИОМИОПАТИИ В НОЗОЛОГИИ ПОВРЕЖДЕНИЙ МИОКАРДА
Кардиомиопатии (от греч. kardia -сердце, mys, myos - мышца и pathos - страдание, болезнь) - группа болезней сердца, общим для которых является избирательное поражение миокарда. Этот термин был предложен W. Bridgen в 1957 году, а алкогольное поражение сердца впервые было описано еще в XIX веке. Оно было характерно для жителей Мюнхена, выпивавших в год около 430 литров пива.
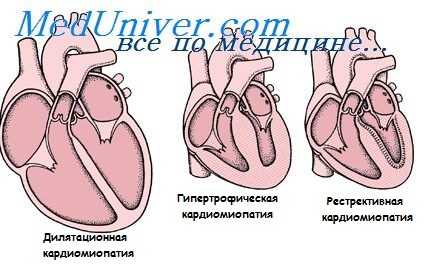
В настоящее время экспертами ВОЗ принята следующая классификация кардиомиопатий (1995):
- дилатационные;
- гипертрофические;
- рестриктивные;
- аритмогенный правый желудочек;
- неклассифицируемые.
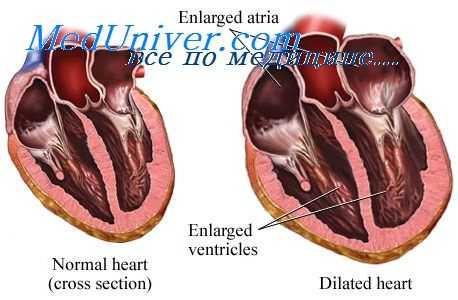
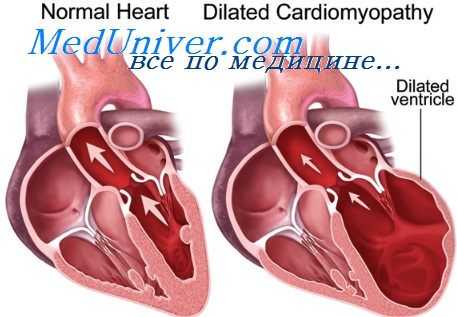
АКМП - приводящая к систолической дисфункции дилатация полостей сердца, развившаяся в связи с избыточным приемом алкоголя. Согласно данной классификации, АКМП относится ко вторичным токсическим дилатационным кардиомиопатиям (ДКМП). Отметим, что по оценкам разных авторов в 2-36% случаев дилатационная кардиомиопатия имеет алкогольную этиологию, в этом случае ее следует рассматривать в рамках АКМП (2,32,42).
МОРФОЛОГИЯ АЛКОГОЛЬНОЙ КАРДИОМИОПАТИИ
Клинические и гистологические признаки алкогольной и идиопатической дилатационной кардиомиопатий во многом схожи. Тем не менее, установление клинического диагноза «АКМП» имеет большое практическое значение, поскольку это одно из немногих потенциально обратимых состояний, ассоциированных с сердечной недостаточностью (СН), при котором полный отказ от алкоголя обыкновенно приводит к значительному улучшению насосной функции левого желудочка (ЛЖ).
При АКМП макроскопически отмечается расширение всех полостей сердца при минимальной неравномерной гипертрофии миокарда. По мере усугубления СН дилатация прогрессирует. На секции обращает на себя внимание обычно массивное субэпикардиальное скопление жировой клетчатки; ввиду жировой дистрофии миокард может приобретать желтоватый оттенок. Важно отметить, что коронарные артерии при АКМП, как правило, остаются интактными или имеют довольно умеренные атеросклеротические поражения.
Гистологически АКМП характеризуется прежде всего вакуолизацией и мелкокапельной жировой дистрофией саркоплазмы большинства кардиомиоцитов. Также определяется внеклеточное ожирение в строме миокарда и вокруг интрамуральных сосудов. В одном поле может наблюдаться сочетание гипертрофии и атрофии кардиомиоцитов. Содержание липофусцина повышено, зерна пигмента распространяются от полюсов по всей саркоплазме. В миокарде могут встречаться небольшие круглоклеточные скопления, формирующиеся как реакция на повреждение. Миофибриллы беспорядочно расположены в этих кардиомиоцитах.
Нередко наблюдаются утолщение отдельных участков эндокарда по типу фиброэластоза, выраженный интерстициальный и периваскулярный фиброз всего миокарда. При ДКМП соединительная ткань локализуется преимущественно во внутренней трети миокарда.
Некоторые авторы обнаруживают очаговую лимфогистиоцитарную инфильтрацию миокарда с примесью плазматических клеток и эозинофилов, а также признаки аллергического поражения сосудов (32).
При электронной микроскопии впечатляет реорганизация митохондрий - органелл-мишеней этанола. На ранних стадиях происходит пространственная реорганизация митохондриального ретикулума: межмитохондриальные контакты исчезают, и митохондрии образуют отдельные кластеры, равномерно распределенные по миоцитам. На поздних стадиях происходят необратимые деструктивные изменения ультраструктуры митохондрий. Появляются гигантские и разделенные мембраной митохондрии. Внутри митохондрий появляется множество липофусциновых гранул - предшественников саркоплазматических липофусциновых гранул. При прогрессировании данного заболевания формируются межмитохондриальные контакты, образованные соединением не только наружных, но и внутренних мембран соседних митохондрий. В целом, популяции митохондрий в кардиомиоцитах присуща гетерогенность (Сударникова Ю.В., 2000).
Характерные изменения можно обнаружить и в других органах. Например, в надпочечниках, где при АКМП увеличивается содержание катехоламинсодержащих структур, что, по-видимому, вызвано увеличением биосинтеза гормонов в железе и задержкой их выделения в кровяное русло. При ДКМП, напротив, отмечается количественное уменьшение катехоламинсодержащих в медулле, что, вероятно, обусловлено, с одной стороны, ослаблением синтетической активности хромаффиноцитов, с другой - повышенным выбросом гормонов в кровь (28).
ПАТОФИЗИОЛОГИЯ АЛКОГОЛЬНОЙ КАРДИОМИОПАТИИ
Структура неразрывно связана с функцией. Каким же образом формируются описанные изменения, клиническим выражением которых являются дисфункция желудочков сердца и снижение сократимости миокарда?
Главным этиологическим фактором развития алкогольной кардиомиопатии является хронический избыточный прием алкоголя. По оценкам разных авторов, истинная АКМП обычно развивается при потреблении алкоголя в количестве, эквивалентном ≈100 мл чистого этанола, ежедневно в течение 10-20 лет. Эти цифры могут показаться внушительными, и здесь необходимо вспомнить о том, что среднее потребление алкоголя в России на душу населения колеблется около отметки 15 л, что соответствует 40 мл чистого этанола в день. 82% взрослого мужского населения России являются потребителями спиртного, а каждый пятый из этого множества постоянно злоупотребляет алкоголем (12, 22, 24).
ВОЗМОЖНЫЕ МЕХАНИЗМЫ ТОКСИЧЕСКОГО ДЕЙСТВИЯ АЛКОГОЛЯ НА КАРДИОМИОЦИТЫ
Эффекты алкоголя представляются дозозависимыми: рост вероятности развития ИБС отмечают как при полном воздержании от алкоголя, так и при злоупотреблении им (19). Очертим круг возможных механизмов кардиомиопатогенного действия этанола.
Наиболее важными из них представляются следующие:
- влияние на метаболизм и энергообеспечение клетки;
- прямое токсическое действие ацетальдегида и этанола на синтез белка;
- срыв сопряжения между возбуждением и сокращением;
- свободнорадикальное повреждение;
- нарушение липидного обмена;
- дисбаланс катехоламинов и других гормонов;
- ионный дисбаланс;
- воздействие на цитоскелет;
- активация провирусов;
- изменение процессов возбуждения и проведения в сердечной мышце;
- токсическое действие спиртных напитков, обусловленное примесями металлов (например, кобальта).
- Часть «алкогольного» ацетил-SKoA не окисляется в цикле Кребса, а используется для синтеза липидов (в печени). В миокарде же сходным образом ингибируется окисление липидов, что и приводит к жировой дистрофии.
- Восстановленные интермедиаты начинают окислятся не на первом комплексе ЦПЭ в митохондриях (НАДН-дегидрогеназа ингибирована), а на втором, НАД-независимом, коим является сукцинатдегидрогеназа. Окисление глюкозы и гликогенолиз также ингибируются, морфологическим эквивалентом чего являются значительные скопления гликогена внутри вакуолей кардиомиоцитов.
Показано, что этанол ингибирует синтез белка в кардиомиоцитах только в летальных концентрациях, в то время как даже низкие концентрации ацетальдегида (сопоставимые с таковыми у больных АКМП) значимо угнетают его (18). Кроме того, ацетальдегид опосредованно оказывает положительный хроноионтропный эффект на кардиомиоциты. Он, по всей видимости, достигается за счет повышения высвобождения норадреналина из симпатических нервных волокон.
Как уже отмечалось, при АКМП в надпочечниках накапливаются избыточные количества катехоламинов. Таким образом, характерная для АКМП дисфункция миокарда развивается при двойном дисбалансе катехоламинов: нейромедиаторов в синаптической щели (норадреналин) и гормонов в крови, омывающей миокард (адреналин).
Интересно, что назначение в эксперименте пропранолола с целью нивелирования излишнего влияния катехоламинов, как и предполагалось, сводит на нет положительные инотропный и хронотропный эффекты ацетальдегида, но не влияет на снижение синтеза белка! Значит, ингибирование синтеза белка ацетальдегидом опосредуется другим, отличным от катехоламинового, механизмом (3).
Нарушение сопряжения между возбуждением и сокращением. В 1990 году Guarnieri et al. в серии блестящих экспериментов на изолированной сосочковой мышце, перфузируемой раствором этанола, доказали, что алкоголь вмешивается в мышечное сокращение и нарушает его. Критическим этапом здесь представляется взаимодействие между Са 2+ и миофибриллами. Кроме того, алкоголь в высоких концентрациях проявляет себя как антагонист Са 2+ и Na + -каналов. После прекращения подачи этанола к кардиомиоцитам данные эффекты исчезали. Авторы предположили, что увеличение концентрации Са 2+ в цитозоле кардиомиоцитов может улучшить сократительную функцию кардиомиоцитов.
РАЗВИТИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
Освещенные выше механизмы, реализуя свое пагубное действие на сердце, приводят, в конечном итоге, к развитию СН. Принципиально важным процессом является ремоделирование сердца. Это понятие включает в себя: нарушение структуры сократительного аппарата кардиомиоцитов, их функциональную асимметрию, изменение межклеточных взаимодействий, интерстициальный фиброз, деспирализацию хода мышечных пучков и изменение формы полостей сердца. Употребление термина «ремоделирование» в данном аспекте спорно, так как он скорее указывает на обратное развитие патологических изменений, нежели на их формирование. Возможно, такое понятие, как «демоделирование сердца», более соответствующее приведенной дефиниции, войдет в кардиологический актив, при этом процессы, происходящие в миокарде при назначении (ß-блокаторов и иАПФ, можно будет оправданно называть ремоделированием.
Субклинически на стадии формирования интерстициального фиброза увеличивается ригидность стенки желудочка. Конечно-диастолическое (КД) давление растет на фоне снижения КД объема. Именно так формируется диастолическая дисфункция.
В дальнейшем, при АКМП на передний план выходит систолическая дисфункция миокарда, которая и приводит к СН.
Параллельно этому происходит растягивание фиброзных колец клапанов сердца, обусловленное дилатацией желудочков. В первую очередь это касается митрального клапана. Следствие данного процесса - клапанная регургитация, которая приводит к дилатации полостей предсердий, а впоследствии - к легочной гипертензии. Последняя, однако, редко достигает высокой степени при АКМП.
В настоящее время в России диагноз «АКМП» может быть установлен кардиологом без психиатрической консультации больного, поэтому четкие критерии данного состояния особенно востребованы.
Диагностика АКМП сложна и должна включать в себя непосредственное обследование больного, сбор алкогольного анамнеза, лабораторные и инструментальные методы исследования. Следует учитывать, что клиническая картина АКМП сходна с ДКМП.
Типичный больной АКМП - мужчина 30-55 лет, жалующийся на боли в сердце, одышку, с признаками избыточного потребления алкоголя без сопутствующей патологии печени.
Боль при АКМП обычно появляется на следующий день после алкогольного эксцесса, а чаще - после нескольких дней злоупотребления алкоголем. При этом она локализуется в области соска, верхушки сердца, иногда захватывая всю предсердечную область. Обычно боль проявляется постепенно, исподволь, бывает ноющей, тянущей, колющей. Для нее нехарактерны приступообразность, связь с физической нагрузкой и купирование приемом нитратов. На Эхо-КГ при АКМП, в отличие от ИБС, наблюдается дилатация полостей сердца со снижением сократимости миокарда, не имеющим очагового характера.
Объективным методом, помогающим в диагностике кардиомиопатии и дифференциальной диагностике АКМП и ДКМП, является прижизненная субэндокардиальная биопсия миокарда с морфологическим исследованием.
СТРАТЕГИЯ ЛЕЧЕНИЯ АЛКОГОЛЬНОЙ КАРДИОМИОПАТИИ
Терапия АКМП может быть признана адекватной, только если она проводится по трем следующим направлениям:
- предотвращение эпизодов приема алкоголя;
- профилактика и лечение СН;
- коррекция метаболических нарушений, вызвавших АКМП.
Стандартное лечение СН при АКМП само по себе мало отличается от лечения СН другой этиологии и подробно рассмотрено в соответствующих руководствах. В данной работе считаем важным обратить внимание на перспективы метаболической терапии АКМП и сопутствующей СН.
Именно метаболическая терапия является патогенетически обоснованным методом лечения АКМП как заболевания, развившегося именно вследствие патологических метаболических сдвигов в биохимическом континууме кардиомиоцитов. Наиболее многообещающими в этом отношении представляется использование лекарственных средств с цитопротективным эффектом: триметазидин, левокарнитин, фосфокреатин и др. Восстановление потенциально обратимо нарушенного метаболизма возможно при таком назначении препаратов, чтобы каждый предыдущий создавал субстраты для действия последующего медикамента.
В такой схеме триметазидин работает на первом рубеже. Он переключает метаболизм миокарда с использования жирных кислот на аэробный распад глюкозы (как более эффективный путь), ингибируя митохондриальную кетоацил-СоА-тиолазу. Таким образом, цикл Кребса «стимулируется» вступающим в него дополнительным пируватом.
Новинкой фармацевтического рынка является препарат Ангиозил ® ретард - высококачественный препарат триметазидина.
Ангиозил ® ретард выпускается в таблетках с контролируемым высвобождением, что, на настоящий момент, является самой современной и востребованной лекарственной формой, позволяющей поддерживать фармакологически активную концентрацию триметазидина в крови в течение длительного времени.
Перспектива использования именно триметазидина (Ангиозил ® ретард) в лечении АКМП делает реально достижимой главную цель метаболической терапии - ускорение всей цепочки реакций от окисления глюкозы и жирных кислот до фосфорилирования актина. Такой подход может быть эффективным как в повышении фракции выброса, так и в обращении вспять дистрофических изменений. Степень приверженности к терапии, как известно, тесно коррелирует с кратностью приема препарата. По этой причине все чаще выбор падает на ретардированные формы триметазидина (Ангиозил ® ретард), который применяется 2 раза в сутки, что удобно для пациента.
В России экономические аспекты терапии приобретают особое значение. Стоимость препарата, особенно, когда речь идет о необходимости проведения комбинированного лечения, во многом определяет выбор всей схемы лечения АКМП, как и любых других нозологий.
Поэтому все чаще врачи отдают предпочтение качественным дженерическим препаратам, которые подтвердили свою эффективность, хорошую переносимость и при этом доступным для пациентов.
Ангиозил ® ретард является высококачественным дженерическим препаратом триметазидина российского производства с подтвержденной биоэквивалентностью с более дорогостоящим оригинальным препаратом.
Сумма этих качеств позволяет препарату Ангиозил ® ретард быть современным средством эффективной цитопротекции, при этом оставаясь доступным самому широкому кругу пациентов.
Таким образом, АКМП в настоящее время является значимой проблемой, особенно в России. Хотя определенные звенья патогенеза данного заболевания выяснены, полное понимание связи между этиологией и клиникой еще предстоит обрести.
Еще одной важной задачей является разработка четких схем лечения АКМП, которые позволили бы достичь значимого клинического успеха в лечении этой патологии.
Роль ретардированных триметазидинов (Ангиозил ® ретард) в этом процессе трудно переоценить.
В идеале, хорошо спланированное рандомизированное клиническое исследование с применением метаболической терапии, в первую очередь триметазидина ретард, сможет помочь найти оптимальную схему лечения АКМП.
Нарушения метаболизма миокарда при алкоголизме. Алкогольная гипоксия
До настоящего времени остается дискуссионным вопрос, сам ли этанол или его первый метаболит ацетальдегид оказывают повреждающее действие на миокард. М. Wong (1974) показал, что инфузия 25— 30 мг/кг/мин алкоголя в миокард собаки в течение 15 мин при трансвенозной биопсии приводит к значительному нарастанию концентрации триглицеридов и одновременному снижению работы сердца.
Возможность образования за такой короткий период ацетальдегида, по мнению М. Wong (1974), исключается; к тому же сердце не содержит алкогольдегидрогеназы (A. Whereat и J. Perloff, 1973). В настоящее время принято считать, что этанол оказывает прямое кардиодепрессивное действие, независимо от добавочного действия ацетальдегида, который повреждает миокард аналогичным образом.
Еще раньше Т. Regan с соавторами (1964), инфузируя 15% раствор этанола интактным собакам с тем, чтобы плазменный уровень его составлял 150—215 мг%, обнаружил при исследовании крови из коронарного синуса снижение усвоения главного сердечного субстрата — свободных жирных кислот, нарастание экстракции триглицеридов и накопление их в миокарде, значительную утечку ионов калия и фосфора, а также глютаминощавелевоуксусной трансаминазы.
Параллельно этим изменениям уменьшались ударный объем и сила сокращений левого желудочка, повышалось конечное диастолическое давление. Исследования были повторены на собаках, получавших этанол на фоне полноценного, содержащего в необходимом количестве белки и витамины питания в течение двух лет, причем и здесь наблюдался сдвиг инкорпорации жиров левым желудочком от фосфолипидов к триглицеридам; отмечалось также увеличение содержания молочной кислоты, отрицательный баланс изоцитрат- и лактатдегидрогеназы, что свидетельствовало о повреждении клеточных мембран и митохондрий.
Дальнейшие исследования на животных в хроническом эксперименте выявили снижение активности изоцитратдегидрогеназы и пируватдегидрогеназы митохондрий, уменьшение способности саркоплазматического ретикулума связывать и выделять кальций, снижение концентрации в нем эндогенного кальция. Эти изменения сопровождались угнетением дыхания митохондрий.
Изучая метаболические сдвиги в сердечной мышце крыс, А. Гвоздяк с соавторами (1976) пришли к выводу, что хроническая алкогольная интоксикация вызывает торможение окислительных процессов, снижает энергетический резерв сердечной мышцы, что в свою очередь отражается на сократительной способности миофибрилл и представляет собой основу для развития сердечной недостаточности при алкогольной кардиомиопатии.
В. И. Шишов с соавторами (1977) показали, что острая интоксикация алкоголем вызывает снижение активности глутаматдегидрогеназы, малатдегидрогеназы, НАДН : цитохром-с-оксидазы и цитохром-с-оксидазы в миокарде крыс при одновременном повышении активности сукцинат- и лактатдегидрогеназы, увеличении концентрации пирувата, сукцината, альфа-кетоглутарата и снижении концентрации щавелевоуксусной кислоты.
Длительное введение алкоголя способствует интенсификации гликолиза и НАДН : цитохром-с-оксидоредуктазного пути с преимущественным окислением сукцината и увеличением активности цитохром-с-оксидазы, что рассматривается авторами как приспособительная реакция миокарда на хроническую «алкогольную гипоксию».
Читайте также: