Врожденные аномалии почек. Агенезия почек
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
К врождённой патологии почек относят пороки развития и метаболические нефропатии.
Пороки развития связаны с нарушением эмбриогенеза и выражаются различными аномалиями.
Разновидности
Аномалии расположения (почки располагаются в грудной клетке, подвздошной области, тазовой полости и др.), взаимоотношения и формы (сращенные почки, например подковообразная) клинически проявляются, как правило, болью, возникающей при перемене положения тела, физическом напряжении. Лечение преимущественно консервативное. Оперативное лечение показано при осложнениях (почечно-каменной болезни, гнойном пиелонефрите и гидронефрозе).
Аномалии количества почек наиболее часто встречаются в форме удвоения почек. Паренхима удвоенной почки имеет признаки гипоплазии, дисплазии, что способствует развитию различных заболеваний, в первую очередь пиелонефрита. При полной анатомической и функциональной неполноценности одной или обеих добавочных почек производят их удаление.
Аномалии почечной паренхимы и чашечно-лоханочной системы
Агенезия почки — результат отсутствия закладки органа в процессе эмбриогенеза. Дети с двусторонней агенезией почки обычно рождаются мертвыми. При односторонней агенезии почки единственная почка обычно гипертрофирована и полноценна в функциональном отношении.
Аплазия почки — тяжелая степень недоразвития почечной паренхимы, нередко сочетающаяся с отсутствием мочеточника. Обычно аплазия почки не проявляется клинически и распознается при обследовании по поводу заболеваний противоположной почки. В редких случаях отмечается артериальная гипертензия. Необходимость лечебных мероприятий при аплазии почки возникает при резко выраженных болях, развитии артериальной гипертензии. Лечение оперативное — удаление аплазированной почки.
Гипоплазия почка — уменьшение в размерах почки. Большинство детей с выраженной двусторонней гипоплазией почек погибает в первые годы жизни. Односторонняя гипоплазия почки клинически может быть бессимптомной. Однако в такой почке нередко возникает пиелонефрит, зачастую приводящий к развитию почечной гипертензии. Противоположная почка компенсаторно гипертрофирована (увеличена). Лечение при односторонней гипоплазии, осложненной пиелонефритом и гипертензией, обычно сводится к нефрэктомии.
Кистозные аномалии паренхимы представлены различными по локализации и количеству кист вариантами. Среди них самый частый — поликистоз. Нередко сочетается с кистозными изменениями других органов (печень, селезенка). Симптомы неспецифичны. Возможны тупые боли в пояснице, эритроциты в моче, артериальная гипертензия, ночное учащенное мочеиспускание. При поликистозе почки часто развиваются пиелонефрит, почечно-каменная болезнь, туберкулез почек. Лечение направлено на борьбу с пиелонефритом, гипертензией, нарушениями водно-электролитного баланса. Оперативное вмешательство становится необходимым при почечном кровотечении, обтурирующем (закрывающем просвет) камне, нагноении кист, развитии злокачественной опухоли почек.
Клинически значимыми аномалиями почечных сосудов являются фибромускулярная дисплазия (врожденное недоразвитие мышечной оболочки артерии с замещением ее фиброзной тканью),врожденные стеноз и аневризма почечной артерии, приводящие к развитию вазоренальной артериальной гипертензии. Лечение заключается в протезировании почечной артерии.
Наследственные метаболические нефропатии развиваются при генетически обусловленных нарушениях обмена веществ в организме с вовлечением в патологический процесс почечных механизмов. К ним относят наследственный нефрит, включая синдром Альпорта, различные варианты почечных тубулопатий (синдром Фанкони, фосфат-диабет, первичная оксалурия, цистиноз).
Для наследственных нефропатий, несмотря на их многообразие, характерен ряд общих клинических признаков. Во-первых, наличие однотипных заболеваний в семье, что отчетливо проявляется при сопоставлении родословных, во-вторых, длительное скрытое течение, нередко с "изолированным мочевым синдромом", что чаще расценивается как одна из форм латентного нефрита. Больным с наследственными нефропатиями свойственны множественные внешние «малые аномалии» (диспропорциональные конечности, низкий рост и т.п.). Наконец, отмечается раннее снижение почечных функций.
Лечение проводится в нефрологическом отделении и имеет синдромологический и в определенной мере заместительный характер. При каждом виде нефропатии назначается специальная диета. Чтобы стимулировать синтез ферментов почечных канальцев, ответственных за обмен того или иного вещества назначают витамины, метаболические препараты. Лечение длительные, больные должны находиться под наблюдением нефролога.
Профилактика наследственных нефропатий связана с дальнейшим расширением медико-генетического консультирования. Профилактика прогрессирования наследственных нефропатий включает устранение токсических, аллергизирующих и других неблагоприятных воздействий внешней среды.
Пройти диагностику и лечение заболевания Вы можете в нефрологическом отделении Центра нефрологии и диализа нашей клиники.
Для получения подробной информации и записи на прием обращайтесь к нам по телефону контакт-центра: (495) 925-02-02 (круглосуточно).
Врожденные аномалии почек. Агенезия почек

Врожденные пороки развития почек – это стойкие морфологические изменения, выходящие за пределы вариаций нормы, приводящие к нарушению функции почек.
Врожденные аномалии почек и мочевыводящих путей встречаются с частотой от 3 до 6 новорожденных на 1000 и составляют порядка 20-30% всех аномалий, выявленных в перинатальном периоде.
Аномалии развития почек:
- добавочная почка;
- удвоение почек (полное и неполное).
- гипоплазия почек простая (уменьшение относительной массы органа без нарушения структуры и функции);
- гипоплазия почек диспластическая (уменьшение относительной массы органа с нарушением структуры и функции);
- агенезия – полное отсутствии почек (одно, двусторонняя).
- аномалии положения – дистопии (гомолатеральная дистопия (торакальная, поясничная, подвздошная и тазовая) или гетеролатерильная-перекрестная дистопия);
- нефроптоз;
- аномалии ориентации: ротация почек.
3. Аномалии взаимоотношения и формы (сращенные почки):
- симметричные (подковообразная и галетообразная почки) формы сращения;
- асимметричные (L-, S-, I-образные почки) формы сращения. Асимметричные формы сращения характеризуются соединением почки противоположными полюсами
4. Аномалии чашечно-лоханочной системы:
- мегакаликоз;
- чашечковый дивертикул (к особенностям строения чашечно-лоханочной системы относится так же пиелоэктазия).
Аномалии почечных сосудов:
1. Аномалии артерий:
- полное отсутствие;
- гипоплазия почечной артерии;
- двойная почечная артерия;
- множественные почечные артерии;
- стеноз почечной артерии;
- аневризма почечной артерии;
- фибромускулярная дисплазия.
- кольцевидная почечная вена;
- ретроаортальная левая почечная вена.
3. Артериовенозные свищи.
4. Аномалии лимфатических сосудов.
5. Нарушение иннервации органов мочевой системы нередко с синдромом нейрогенного мочевого пузыря.
Аномалии дифференцировки (структуры) почек:
- бескистозные формы дисплазии;
- кистозная дисплазия почек.
- тотальный поликистоз почек;
- кортикальный поликистоз почек;
- микрокистоз коры;
- поликистоз пирамид;
- поликистоз почек неклассифицированный.
Тубулопатии (первичные и вторичные тубулопатии):
1. Наследственный нефрит:
- синдром Альпорта;
- нефрит семейный хронический без глухоты;
- нефрит с полинейропатией;
- семейная доброкачесвенная гематурия.
2. Эмбриональная опухоль почек (опухоль Вильмса).
Пороки развития почек могут иметь клинические проявления (боль, дизурия, развитие воспаления), но могут быть и случайной диагностической «находкой». Крупные пороки развития почек могут приводить как к летальному исходу, так и к стойкому нарушению почечной функции.
Учитывая разнообразие и распространенность врожденных пороков развития почек, целесообразно планово, в возрасте 1 месяца, выполнить ребенку УЗИ почек. Возможно заподозрить наличие ряда пороков мочевой системы ребенка уже при ультразвуковом обследовании беременной. В этом случае после рождения ребенка необходимо выполнить малышу УЗИ почек и УЗИ мочевого пузыря, а так же записаться на прием к детскому нефрологу.
Почечные аномалии
Заболеваемость на аутосомно-рецессивную поликистозную болезнь почек составляет около 1/10 000– 1/20 000 новорожденных; болезнь вызвана мутацией гена PKHD1, который расположен на хромосоме 6p21. Аутосомно-доминантный поликистоз почек Аутосомно-доминантная поликистозная болезнь почек (АДПБП) Поликистозная болезнь почек (ПБП) – это наследственное заболевание с формированием почечных кист, вызывающее постепенное увеличение обеих почек, иногда прогрессирующее до почечной недостаточности. Прочитайте дополнительные сведенияАутосомно-рецессивная поликистозная болезнь почек поражает
Почки, как правило, значительно увеличены и содержат небольшие кисты; почечная недостаточность обычно формируется в детском возрасте.
Печень увеличена, характерны перипортальный фиброз, пролиферация желчных протоков и рассеянные кисты; оставшаяся паренхима печени является нормальной. Фиброз вызывает портальную гипертензию в возрасте 5–10 лет, но функция печени нормальная или с незначительными нарушениями.
Тяжесть заболевания и его прогрессирование варьируются. Тяжелый вариант течения может манифестировать пренатально, вскоре после рождения или в раннем детском возрасте ренальными симптомами; менее тяжелые варианты течения проявляются в более старшем или подростковом возрасте и чаще с печеночными симптомами.
Новорожденные с данной патологией имеют выпуклый живот с огромными упругими гладкими симметричными почками. У новорожденных с тяжелым повреждением почек обычно развивается гипоплазия легких, вторичная по отношению к симптомам внутриутробного нарушения функции почек и маловодия.
У пациентов в возрасте 5–10 лет возникают признаки портальной гипертензии Портальная гипертензия Портальная гипертензия – повышенное давление в воротной вене. Наиболее частыми причинами этого состояния являются цирроз (в развитых странах), шистосоматоз (в эндемических областях) или сосудистые. Прочитайте дополнительные сведения , такие как варикозное расширение вен пищевода и желудка и гиперспленизм. У пациентов в подростковом возрасте нефромегалия менее заметна, почечная недостаточность может быть от легкой до умеренной, и основные симптомы связаны с портальной гипертензией.
Диагностика аутосомно-рецессивной поликистозной болезни почек может быть затруднительной, особенно без семейного анамнеза. УЗИ может показать наличие почечных или печеночных кист; для постановки окончательного диагноза может потребоваться биопсия. УЗИ на поздних сроках беременности обычно позволяет предположить диагноз уже внутриутробно. Если послеродовое УЗИ не однозначно, МРТ или КТ могут быть диагностически значимыми. При необходимости, когда клинические критерии не соответствуют, возможно проведение молекулярного теста гена PKHD1.
Многие новорожденные умирают в первые несколько дней или недель жизни от легочной недостаточности. У большинства выживших детей развивается прогрессирующая почечная недостаточность, часто требующая заместительной терапии. Опыт трансплантации почки с трансплантацией печени или без нее ограничен. После проведения трансплантации нужно контролировать гиперспленизм Гиперспленизм Гиперспленизм – это цитопенический синдром, обусловленный спленомегалией. (См. также Обзор селезенки (Overview of the Spleen)). Гиперспленизм является вторичным процессом, который может возникнуть. Прочитайте дополнительные сведения , чтобы устранить трудности с гиперспленизм-индуцированной лейкопенией, увеличивающей риск системной инфекции. Портальную гипертензию можно лечить с помощью установки портокавального или сплено-ренального шунтов, снижающих заболеваемость, но не смертность.
Аномалии дублирования
Дополнительные системы сбора могут быть односторонними или двусторонними и могут включать почечную лоханку и мочеточник (дополнительная почечная лоханка, двойная или тройная лоханка и мочеточник), чашечку или устья мочеточника. Удвоенная почка имеет одну почечную единицу с более чем одной собирательной системой. Эта аномалия отличается от сросшихся почек тем, что предполагает слияние двух почечных паренхиматозных единиц с отдельными собирательными системами. Некоторые аномалии дублирования имеют эктопию мочеточника с/без уретероцеле Уретероцеле Аномалии мочеточников часто встречаются вместе с почечными аномалиями, но могут развиваться независимо. Осложнения включают: Обструкцию, пузырно-мочеточниковый рефлюкс, инфекцию и формирование. Прочитайте дополнительные сведения и/или везикоуретерального рефлюкса Везикоуретеральный рефлюкс (ВУР) Пузырно-мочеточниковый рефлюкс (ПМР) является ретроградным забросом мочи из мочевого пузыря обратно в мочеточник и собирательную систему. Рефлюкс предрасполагает к инфекции мочевыводящих путей. Прочитайте дополнительные сведения (ВУР).
Ведение зависит от анатомии и функционировании каждого отдельно дренируемого сегмента. Хирургическая коррекция может быть необходима для устранения обструкции или ВУР.
Аномалии слияния
Подковообразная почка, наиболее распространенная аномалия слияния, возникает при объединении почечной паренхимы с каждой стороны позвоночного столба в соответствующих (как правило) нижних полюсах; перешеек почечной паренхимы или фиброзной ткани соединяется по средней линии. Мочеточники располагаются медиально и спереди от этого перешейка, в основном, функционируют хорошо. Обструкция, если она присутствует, как правило, является вторичной по отношению к впадению мочеточников высоко в почечную лоханку. Пиелопластика устраняет обструкцию и может быть выполнена без резекции перешейка.
Перекрестная почечная эктопия является второй наиболее распространенной аномалией слияния. Почечная паренхима (представляющая обе почки) находится с одной стороны позвоночника. Один из мочеточников пересекает срединную линию и впадает в мочевой пузырь со стороны, противоположной сросшимся почкам. При обструкции лоханочно-мочеточникового сегмента методом выбора лечения является пиелопластика.
Галетообразная почка развивается гораздо реже. При тазовой дистопии единственной почки она обслуживается двумя собирательными системами и мочеточниками. При наличии обструкции необходима реконструкция.
Мальротация
Мальротации, как правило, имеют небольшое клиническое значение. На УЗИ часто диагностируют гидронефроз. В случае, если врачи подозревают возможную обструкцию, может быть проведена магнитно-резонансная урография или КТ почек.
Мультикистозная дисплазия почки (МКДП)
В этом случае нефункционирующая часть почки состоит из несообщающихся кист, между которыми находятся твердая ткань, состоящая из фиброза, включений хрящей, а также очагов канальцев. Как правило, выявляют также атрезию мочеточника. Противоположная почка, как правило, нормальная, но до 10% пациентов могут иметь пузырно-мочеточниковый рефлюкс Везикоуретеральный рефлюкс (ВУР) Пузырно-мочеточниковый рефлюкс (ПМР) является ретроградным забросом мочи из мочевого пузыря обратно в мочеточник и собирательную систему. Рефлюкс предрасполагает к инфекции мочевыводящих путей. Прочитайте дополнительные сведения или обструкцию мочеточникового сегмента. Часто почки подвергаются прогрессивной инволюции и, в конечном счете, больше не визуализируются на УЗИ. Развитие новообразований, инфекций, и/или артериальной гипертензии встречаются редко.
Большинство экспертов рекомендуют наблюдение для контроля инволюции. Нефрэктомию можно рассматривать при наличии твердой ткани, прогрессивного расширения, или, в редких случаях, гипертензии или разрыва кисты, которая вызывает боль.
Почечная агенезия
Двусторонняя агенезия почек как часть синдрома маловодия, гипоплазии легких, конечностей и лицевых аномалий (классический синдром Поттера) приводит к летальному исходу в течение от нескольких минут до нескольких часов после рождения. Гибель плода является распространенным явлением.
Односторонняя почечная агенезия составляет около 5% почечных аномалий. Многие случаи являются результатом полной внутриутробной инволюции мультикистозной диспластической почки. Обычно она сопровождается агенезией мочеточника с отсутствием ипсилатерального треугольника и устья мочеточника. Тем не менее, надпочечник, находящийся на стороне поражения, не вовлекается в процесс. Лечение не требуется; компенсаторная гипертрофия единственной почки поддерживает нормальную почечную функцию. Поскольку почки имеют общее эмбриональное происхождение с семявыводящими протоками и маткой, мальчики могут иметь отсутствие семявыводящих протоков, а девочки аномалии матки.
Дисплазия почек
При дисплазии почек (гистологический диагноз) почечная сосудистая сеть, канальцы, собирательная система или дренажный аппарат развиваются ненормально. Диагноз почечной дисплазии ставится на основании данных биопсии.
При сегментарной форме дисплазии часто лечение почечной дисплазии не требуется. При обширной дисплазии нарушение функции почек может потребовать нефрологической помощи, включая заместительную почечную терапию.
Эктопия почек
Эктопия почек (аномальное расположение почек) обычно развивается, когда почки не могут подняться из места их формирования в малом тазу; редкое исключение – чрезмерно поднявшаяся почка (торакальная эктопия). Тазовая эктопия увеличивает частоту обструкции лоханочно-мочеточникового сегмента, пузырно-мочеточникового рефлюкса Везикоуретеральный рефлюкс (ВУР) Пузырно-мочеточниковый рефлюкс (ПМР) является ретроградным забросом мочи из мочевого пузыря обратно в мочеточник и собирательную систему. Рефлюкс предрасполагает к инфекции мочевыводящих путей. Прочитайте дополнительные сведения и поликистозной дисплазии почек.
Обструкция и рефлюкс тяжелой степени могут быть скорректированы хирургическим путем при наличии показаний (если они вызывают артериальную гипертензию, рецидивирующие инфекции или задержку роста почек).
Гипоплазия почек
Гипоплазия обычно возникает из-за недостаточного ветвления мочеточникового зародыша, что обусловливает формирование недоразвитых маленьких почек с гистологически нормальными нефронами. Если гипоплазия сегментарная, может развиться гипертензия и может потребоваться проведение абляции. Пациенты должны быть обследованы на наличие пузырномочеточникового рефлюкса Везикоуретеральный рефлюкс (ВУР) Пузырно-мочеточниковый рефлюкс (ПМР) является ретроградным забросом мочи из мочевого пузыря обратно в мочеточник и собирательную систему. Рефлюкс предрасполагает к инфекции мочевыводящих путей. Прочитайте дополнительные сведения .
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Журнал «Здоровье ребенка» 6 (49) 2013
Врожденные аномалии количества почек: частота, этиопатогенез, пренатальная диагностика, клиника, физическое развитие, диагностика, лечение и профилактика (часть 1)
Авторы: Никитина Н.А., Старец Е.А., Калашникова Е.А., Галич С.Р., Сочинская Т.В. - Одесский национальный медицинский университет, кафедра пропедевтики педиатрии
Версия для печати
В статье представлены литературные данные о частоте, этиопатогенезе, основных клинических проявлениях, динамике физического развития, современных методах пренатальной и постнатальной диагностики и лечения, а также профилактике врожденных аномалий мочевой системы — агенезии и аплазии почек, удвоения почек и третьей, добавочной почки.
У статті висвітлені літературні дані про частоту, етіопатогенез, основні клінічні прояви, динаміку фізичного розвитку, сучасні методи пренатальної та постнатальної діагностики і лікування, а також профілактику вроджених аномалій сечової системи — аплазії та гіпоплазії нирок, подвоєння нирок та третьої, додаткової нирки.
The literature data about frequency, etiopathogenesis, the main clinical symptoms, dynamic of physical development, modern methods of prenatal and postnatal diagnosis and treatment, as well as prevention of congenital disorders of urinary system — agenesia and aplasia, duplicated kidney and third, additional kidney is presented in article.
аномалии почек, агенезия, аплазия.
аномалії нирок, агенезія, аплазія.
anomalies of the kidneys, agenesia and aplasia.
Со второй половины XX века отмечается значительное учащение пороков развития, особенно в развитых странах. По данным ВОЗ, они обнаруживаются у 2,5–3 % новорожденных, около 1 % составляют генные болезни, 0,5 % — хромосомные и в среднем 1,5–2 % приходится на долю врожденных пороков развития (ВПР), обусловленных действием неблагоприятных экзогенных и эндогенных факторов. Частота выявления ВПР с возрастом ребенка увеличивается и к концу первого года жизни достигает 5–7 %.
Согласно официальной статистике, в Российской Федерации частота ВПР и наследственных заболеваний (НЗ) среди всех новорожденных составляет 4–5 %, а их доля в структуре младенческой смертности достигает 35–40 %. Ежегодно в РФ на каждую тысячу рождается от 40 до 50 детей с ВПР и НЗ. В Украине ежегодно рождается около 10 тыс. детей с ВПР и НЗ. В целом частота регистрируемых новорожденных с генетическими расстройствами в Украине сопоставима с европейскими показателями.
Причины 40–60 % ВПР неизвестны. Это спорадические дефекты рождения, возникающие случайно и имеющие низкий риск повторения у будущих детей. Для 20–25 % аномалий более вероятны многофакторные причины, отражающие комплексное взаимодействие разных небольших генетических дефектов и факторов риска окружающей среды. Остальные 10–13 % аномалий связывают с воздействием внешней среды, для 12–25 % аномалий доказано наличие исключительно генетических причин.
Формирование пороков развития происходит преимущественно в период эмбрионального морфогенеза (3–10я недели беременности) в результате нарушения процессов размножения, миграции, дифференциации и гибели клеток. Эти процессы происходят на внутриклеточном, экстраклеточном, тканевом, межтканевом, органном и межорганном уровнях.
При этом может быть недоразвитие органов либо их частей (гипоплазия) или избыточное их развитие, отсутствие органов либо части тела (агенезия), неправильное положение либо перемещение органов (дистопия), неправильное формирование той или иной ткани (дисплазия). Различают двойные (множественные) пороки развития, в основе которых лежат нарушения развития двух и более органов, и одиночные, связанные с нарушением формообразования одного органа.
Врожденные пороки мочевой системы относятся к наиболее частым аномалиям, составляя 35–40 % от пороков всех органов и систем.
Большое разнообразие врожденных аномалий развития систематизировано в классификации А.В. Люлько с соавт. (1984), в которой к аномалиям количества относятся:
— агенезия одной или обеих почек;
— третья, добавочная почка;
— удвоение почек одно и двустороннее.
Агенезия почки (почек) — полное отсутствие почечных структур одной или обеих почек. Агенезия почек может сочетаться с одно или двусторонним отсутствием мочеточников и мочевого пузыря.
Согласно МКБ10 выделяют Q60 Агенезия и другие редукционные дефекты почки. Включает атрофию почки врожденную и инфильтративную, врожденное отсутствие почки.
Q60.0 Агенезия почечная односторонняя.
Q60.1 Агенезия почечная двусторонняя.
Q60.2 Агенезия почечная неуточненная.
Q60.3 Гипоплазия почечная односторонняя.
Q60.4 Гипоплазия почечная двусторонняя.
Q60.5 Гипоплазия почечная неуточненная.
Q60.6 Синдром Поттера.
По МКБ10, почечная гипоплазия является одним из видов агенезии. Хотя большинство авторов указывают на существенную разницу между этими пороками: агенезия почки относится к порокам количества, а гипоплазия — к порокам структуры почки. Между этими двумя видами аномалий авторы выделяют аплазию — резкое недоразвитие почки, при котором некоторые почечные структуры сохранены, т.е. аплазия является крайней степенью гипоплазии. Впервые аплазию почки описал Везалий в 1543 г.
Частота. Средняя частота односторонней агенезии, включая случаи аплазии, составляет 1 на 900–1500 рождений, двусторонняя агенезия встречается с частотой 3,5 на 10 000 рождений. Среди всех больных с аномалиями почек аплазия одной почки встречается относительно часто — у 4–8 %. Как правило, отсутствует левая почка. Чаще поражаются мальчики: соотношение полов м : ж при двусторонней агенезии почек — 2,6 : 1, при односторонней — 2 : 1.
Этиология и патогенез. Этиологически данный порок неоднороден. В большинстве случаев агенезия и гипоплазия почек встречаются спорадически. Отмечено увеличение случаев данного порока развития среди детей матерей, страдающих диабетом. Изолированная агенезия почки — следствие нарушения дифференцировки нефрогенной бластемы. А.В. Айвазян с соавт. (1988) относят агенезию (аплазию) почек к эмбриофетопатии первых 6 недель внутриутробного периода, а М.С. Игнатова (1989) — к бластопатии первых 15 дней имплантации, выделяя критический период с 7го по 12й день. Аплазия почки в сочетании с агенезией половых органов возникают вследствие отсутствия дифференцировки нефрогенного края. Тератотропный период (ТТП) такой формы порока — до 24–26го дня эмбриональной жизни. Арения в сочетании с полным или частичным удвоением матки и влагалища возникают в результате нарушения дифференцировки мезонефрогенного протока и уретральной трубки. ТТП данной формы порока — до 6й недели эмбриональной жизни. При двусторонней агенезии почек вследствие нарушения у плода экскреции жидкости развивается маловодие, что, в свою очередь, приводит к компрессии плода. Это и является основным патогенетическим звеном в формировании характерной клинической картины агенезии почек.
Пренатальная диагностика. Срок наиболее ранней пренатальной ультразвуковой диагностики агенезии почек — 13–17 нед. беременности. Выделена патогномоничная триада эхографических признаков агенезии обеих почек (рис. 1): отсутствие изображения структур почек плода в обычном месте и в местах возможной эктопической локализации, отсутствие эхотени мочевого пузыря, маловодие в ранние сроки (до 17 нед. беременности).

Пренатальный диагноз агенезии обеих почек может вызывать серьезные трудности, так как в условиях выраженного маловодия значительно затруднена визуализация внутренних органов плода. По данным разных исследователей, агенезию почек удается точно диагностировать при УЗИ плода в 69–73 % случаев.
Ранним признаком двусторонней агенезии почек является отсутствие мочевого пузыря после 13й нед. гестации. К поздним проявлениям относится маловодие после 16–18й нед., а в некоторых случаях — после 26й нед.
Дифференциальная диагностика проводится с выраженной гипоплазией почек, при которой также отмечается маловодие и может не визуализироваться мочевой пузырь. Важным диагностическим критерием порока является отсутствие почечных артерий (рис. 2б). В норме почечные артерии четко визуализируются при продольном сканировании туловища плода и отходят от аорты (рис. 2а).

Пренатально односторонняя почечная агенезия выявляется значительно реже, чем встречается. Это обусловлено тем, что при односторонней почечной агенезии, как правило, сохраняется нормальное количество амниотической жидкости, визуализируется мочевой пузырь, а надпочечник может быть принят за почку. Может выявляться компенсаторное увеличение имеющейся единственной почки.
Тактика ведения беременности и родоразрешения. При диагностике маловодия, в особенности на ранних сроках беременности, всегда следует осуществлять поиск агенезии почек. При односторонней агенезии рекомендуются пре или постнатальное кариотипирование для исключения хромосомных аномалий и тщательное изучение УЗанатомии внутриутробного ребенка с целью выявления сочетанных пороков.
Двусторонняя агенезия почек сопровождается симметричной формой задержки роста внутриутробного ребенка и маловодием, являющимися клиническими признаками прогрессирующей плацентарной дисфункции. Проведение коррекции плацентарной дисфункции при указанном пороке обычно неэффективно. Высок риск невынашивания, в том числе преждевременных родов и мертворождения, обусловленного внутриутробной гибелью ребенка.
Пренатальное выявление этого летального порока до 22 недель является показанием к прерыванию беременности по медицинским показаниям в поздних сроках. В случаях отказа от прерывания беременности, как и при поздней диагностике порока, используется консервативная акушерская тактика.
Сочетание с другими пороками. Агенезия почек часто сочетается с другими ВПР и является составной частью известных комплексов множественных врожденных пороков развития (синдром каудальной регрессии и др.). При односторонней агенезии почки, как правило, отсутствуют соответствующие мочеточник и половая железа. У девочек агенезия почки в 70 % случаев сочетается с аномалиями половых органов (отсутствие или недоразвитие матки и ее придатков, двурогая матка, атрезия влагалища и др.). У мальчиков в 20 % случаев агенезия почки сочетается с отсутствием или недоразвитием семенного пузырька, предстательной железы и др.
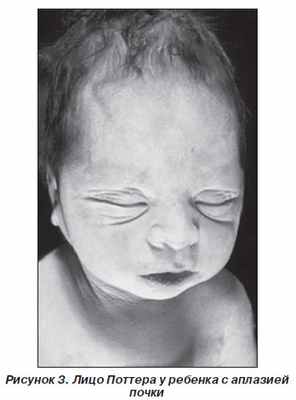
Клиника. Для детей с этой патологией характерны лицевые дисморфии — выступающие лобные бугры, деформированные низко расположенные ушные раковины, широкий плоский нос, микрогнатия, гипертелоризм глаз, эпикант, одутловатость лица — лицо Поттера (рис. 3). Вследствие маловодия и компрессии плода наряду с типичным лицом практически у 100 % детей с данным пороком наблюдаются вторичная гипоплазия легких и деформация нижних конечностей (вывих тазобедренных суставов, косолапость), чрезмерная складчатость кожи и увеличение размера живота. Почти половина детей с агенезией почек рождаются недоношенными. Порок летальный — дети с агенезией (аплазией) обеих почек рождаются мертвыми или умирают в первые дни после рождения, хотя описаны случаи жизни детей без почек в течение 10–23 дней.
Ранним и нередко единственным проявлением агенезии и аплазии почек является анурия. Проявления односторонней агенезии и аплазии отсутствуют. Так как аплазия почки сопровождается обычно гипертрофией контралатеральной, при пальпации живота ее принимают за опухоль. Клиническое значение состоит в опасности заболевания единственной почки. Полное урологическое обследование в данном случае выявляет агенезию или аплазию одной почки (рис. 4).

Ранняя неонатальная и постнатальная диагностика заключается в УЗИ почек новорожденного при наличии множественных стигм дизэмбриогенеза, пороков развития половых органов и длительной (более 12 часов) анурии, а также цистоскопии, экскреторной урографии, аортографии, пневмоперитонеографии.
Лечение. При двусторонней аплазии порок летальный, при нарушении функции единственной почки показана трансплантация органа. В случае односторонней аплазии при наличии здоровой почки прогноз благоприятный. Профилактика заключается в предупреждении воздействия тератогенных факторов в первые 2 недели беременности и в прерывании беременности при обнаружении УЗкартины отсутствия или аплазии обеих почек. При генетическом консультировании семьи обсуждается прогноз и риск повторения порока, возможные варианты наследования и результаты патологоанатомического исследования. В связи с этим рекомендовано УЗИ почек у ближайших родственников. Агенезию почек при аутопсии дифференцируют с выраженной гипоплазией почек (олигонефротическая гипоплазия), которая обычно не имеет семейного повторения.
1. Богатирьова Р.В. Міжрегіональна програма пренатального генетичного моніторингу природжених вад розвитку «Реплікація» // ПАГ. — 1998. — № 5. — С. 6062.
2. Врожденные пороки развития: Практическое руководство / В.Н. Запорожан, И.Л. Бабий, С.Р. Галич, Е.Л. Холодкова, Н.А. Никитина, Е.А. Калашникова. — Одесса: ОНМедУ, 2012. — 320 с.
3. Гельдт В.Г., Кузовлева Г.И. Диагностика пороков мочевыделительной системы у новорожденных и грудных детей // Педиатрия. — 2006. — № 1. — C. 8794.
4. Горин В.С., Серов В.Н. и др. Современные методы пренатальной диагностики хромосомных заболеваний // Вестник. — 2000. — № 3. — С. 4753.
5. Давыденкова Е.Ф., Бутомо И.В. Основные направления профилактики врожденных пороков развития // Педиатрия. — 1985. — № 12. — С. 5153.
6. Зелинская Д.И. О состоянии детской инвалидности и реализации федеральной целевой программы «Детиинвалиды» // Рос. пед. журнал. — 2001. — № 2. — С. 47.
7. Игнатова М.С., Вельтищев Ю.Е. Детская нефрология: Руководство для врачей. — Л.: Медицина, 1989. — 456 с.
8. Калмин О.В., Калмина О.А. Аннотированный перечень аномалий развития органов и частей тела человека: Учебнометодическое пособие. — Пенза: Изд. ПГУ, 2000. — 192 с.
9. Клиническое руководство по ультразвуковой диагностике / Под ред. В.В. Митькова, М.В. Медведева. — М.: Видар, 1997. — Т. 2. — 320 с.
10. Лазюк Г.И. Этиология и патогенез врожденных пороков развития // Тератология человека: Рук. для врачей / Под ред. Г.И. Лазюка. — М.: Медицина, 1991. — C. 1846.
11. Лильин Е.Т., Герасимова О.И., Савицкая Т.В. Перспективы антенатальной профилактической терапии врожденных пороков развития // Тер. архив. — 1990. — Т. 62, № 10. — С. 7778.
12. Лопаткин Н.А., Пугачев А.Г. Детская урология: Руководство. — М.: Медицина, 1986. — 496 с.
13. Лукьянова Е.М. Современные возможности пренатальной диагностики врожденной патологии плода // Перинатологія та педіатрія. — 1991. — Т. 36, № 5. — С. 7477.
14. Майборода Т.А. Пренатальна діагностика вроджених вад розвитку плода // Ультразвукова перинатальна діагностика. — 2000. — № 13. — С. 8793.
15. Минков И.П. Мониторинг врожденных пороков развития: их пренатальная диагностика, роль в патологии у детей и пути профилактики // Перинатологія та педіатрія. — 2000. — № 1. — С. 814.
16. Нікула Е.Т. Частота природженої патології у живонародженних України: Автореф. дис. канд. мед. наук / Укр. наук.гігієн. центр. — К., 1999. — 16 с.
17. Опыт применения УЗисследований для выявления пороков мочевой системы у детей при массовых обследованиях / И.П. Минков, О.Ю. Малютенко, С.А. Крестина, Л.И. Торбинская // Педиатрия. — 1991. — № 5. — С. 8488.
18. Основы практической урологии детского возраста / Люлько А.В., Мурванидзе Д.Д., Возиянов А.Ф. — Киев: Вища шк., 1984. — 286 с.
19. Патологическая анатомия болезней плода и ребенка: Руководство для врачей: В 2 т. / Под ред. Т.Е. Ивановской, Л.В. Леоновой. — М.: Медицина, 1989. — Т. 1. — 384 с.
20. Пороки развития почек и мочеточников / А.В. Айвазян, А.М. ВойноЯсенецкий. — М.: Наука, 1988. — 488 с.
21. Рапопорт С.В., Мотлох Л.Н., Иванова Э.И. Роль врожденных аномалий мочевыводящих путей в развитии заболевания почек у детей // Вопр. охр. мат. и дет. — 1988. — Т. 33, № 10. — С. 6366.
22. Резник Б.Я., Запорожан В.Н., Минков И.П. Врожденные пороки развития у детей. — Одесса: АО «Бахва», 1994. — 448 с.
23. Резник Б.Я., Минков И.П., Подгорная Т.Г., Кривенькая М.Н. и др. Частота и клиникогенетическая характеристика аномалий органов мочевой системы у детей // Урол. и нефрол. — 1991. — № 4. — С. 3741.
24. Рудень В.В. Вдосконалення системи первинної профілактики природжених вад розвитку людини через механізм використання психології її споживача просвітньої інформації // Вісн. наук. досл. — 2000. — № 4. — С. 46.
25. Рудень В.В. Модель управління системою інформаційнопросвітнього забезпечення населення основами медикогенетичних знань з питань попередження виникнення та розвитку природжених вад // Ліки України. — 2000. — № 2(43). — С. 1720.
26. Рудень В.В. Профілактика природжених вад розвитку. — Львів: ЛігаПрес, 2002. — 228 с.
27. Сорокман Т.В., Швигар Л.В. Генетичний моніторинг. Частина І. Проблеми епідеміології уродженних вад розвитку // Здоровье ребенка. — 2007. — № 3(6). — С. 109111.
28. Указ Президента України № 118/99 від 4 лютого 1999 р. «Про цільову комплексну програму генетичного моніторингу в Україні на 1999–2003 роки» // Еженедельник «Аптека». — 1999. — № 6 (15 февраля).
29. Шадлун Д.Р. Шляхи зниження перинатальної смертності на сучасному стані // ПАГ. — 2000. — № 1. — С. 108110.
30. Шейко Л.П. Вроджені вади розвитку у дітей: Автореф. канд. мед. наук. — К., 1998. — 19 с.
31. Ромеро Р., Пилу Д., Дженти Ф. Пренатальная диагностика врожденных пороков развития плода: Пер. с англ. — М.: Медицина, 1997. — 448 с.
32. Zum Bauchdeckena plasiesyndrom / L. Rohden, G. Reppin, I. Iaenecke et al. // Kinderaztl. Prax. — 1980. — Bd. 48, № 12. — S. 640650.
Врожденные аномалии почек и мочеточников
Формирование почек может сопровождаться разными дефектами. Иногда нарушается расположение почек (дистопия), их ориентация (незавершенный поворот почки), они могут срастаться (подковообразная почка), одна почка может вообще отсутствовать (агенезия почки), возможна, наличие добавочных почечных артерий. Когда отсутствуют обе почки, ребенок не выживает (синдром Поттера). Почечная ткань также может развиваться неправильно (аплазия, гипоплазия или дисплазия почки). Например, иногда почка содержит множество кист – диагностируют поликистоз почек.
К возможным аномалиям мочеточников относятся: наличие дополнительных мочеточников, аномальное их расположение, сужение (сужение или заращение мочеточника встречается у 0,6% детей) или расширение. Моча из мочевого пузыря может поступать обратно в аномальные мочеточники, что повышает вероятность развития инфекции в почках (пиелонефрита). Сужение мочеточника затрудняет прохождение мочи из почки к мочевому пузырю, что вызывает увеличение почки (гидронефроз) и ведет к ее повреждению.
Поликистоз почек может долгое время не проявляться, однако кисты сдавливают почечную паренхиму, нарушается ее кровообращение. Поликистоз начинает проявлять после 30 лет: возникает полиурия, гипостенурия, гипертония. При пальпации обнаруживаются увеличенные, плотные, бугристые почки, возможно одновременное кистозное поражение печени. При контрастном рентгенологическом исследовании (пиелографии) обнаруживается типичная картина вытянутых лоханок и чашечек в виде «ножек паука».
Агенезия почек – врожденное отсутствие одной или обеих почек (арения). У таких новорожденных складчатая кожа, одутловатое лицо, низко расположенные ушные раковины, широкий и плоский нос, выступающие лобные бугры. Сочетается с врожденными пороками других органов. Дети нежизнеспособны. Агенезия почек встречается редко.
Гипоплазия почек – врожденное уменьшение массы и объема почек. Может быть одно- и двусторонним.
Дисплазия почек – гипоплазия с одновременным наличием в почках эмбриональных тканей. При двусторонней сильной гипоплазии и дисплазии почек дети нежизнеспособны.
Более легкие аномалии развития – сращение почек (подковообразная почка) и дистопия почки клинически не проявляются и обнаруживаются только при инструментальных методах исследования или при развитии осложнений.
Дистопия – врожденное смещение почки (чаще вниз), может сочетаться с аномальным отхождением мочеточника, наличием добавочных почечных артерий. Дистопия почки также приводит к гидронефрозу, в ней легко образуются камни, развивается почечная гипертония.
Дистопия почки выявляется при рентгенографическом исследовании. От опущения почки ее отличает короткий мочеточник (при опущении мочеточник длинный и изогнутый)
Добавочные почечные артерии чаще встречаются слева, они огибают и сдавливают мочеточник, что приводит к гидронефрозу и почечной гипертонии.
Частые пороки развития мочевыводящих путей – удвоение лоханок и мочеточников; агенезия, атрезия, стеноз мочеточников, эктопия их устьев.
Стенозы мочеточника могут быть одно- и двусторонними. Двусторонняя атрезия – смертельный порок, сопровождается гидронефрозом или дисплазией почки. Мочеточник при этом заканчивается слепо и резко расширен выше атрезии. Может иметь вид фиброзного тяжа.
Клапаны мочеточника чаще всего располагаются в верхней трети и представляют собой складки слизистой с мышечными волокнами. Выявляются они в детском возрасте, так как легко возникает инфицирование почки (пиелонефрит), гидронефроз, образуются камни.
Дивертикул мочеточника – это выпячивание его стенки. Также может быть одно- и двусторонним. Осложняется образованием камней, воспалением, разрывом стенки. В дивертикуле может развиться опухоль.
Дилатация мочеточника – расширение мочеточника с атрофией стенки. В норме диаметр мочеточника у взрослых – 1 см, у новорожденных – 0,6 см. Дилатация чаще всего является следствием обструкции и расположена выше места сужения, однако возможна и первичная дилатация без обструкции.
Гидроуретер – расширение и водянка мочеточника вследствие обструкции. Может сочетаться с гидронефрозом или самостоятельно.
Мегалоуретер – расширение и удлинение мочеточника. Может быть первичным и вторичным. Возникает при пузырно-мочеточниковом рефлюксе или обструкции на различных уровнях, как следствие метаболических, токсических, воспалительных и послеоперационных нарушений.
При мегалоуретере мочеточник значительно удлинен, расширен (диаметр до 2 см и более), он извитой, а стенка его утолщена. Все это выявляется при контрастной рентгенографии.
Дисплазия мочеточника – врожденное нарушение строения мышечной стенки мочеточника, при котором наблюдается резкое расширение мочеточника.
Дисплазия мочеточника прогноз неблагоприятный. Хирургическое лечение оказывается малоэффективным.
Гипоплазия мочеточника – врожденное недоразвитие мочеточника – полное или неполное.
Уретероцеле (внутрипузырная киста мочеточника) – выпячивание в мочевом пузыре стенки мочеточника. Чаще встречается у женщин. Может быть одно- и двусторонним. Чаще всего проявляется везикоуретральным рефлюксом и пиурией (лейкоциты в моче). Наиболее частые осложнения – гидронефроз, пиелонефрит, образование камней.
Пороки развития возникают при воздействии на плод тератогенных факторов во время формирования того или иного отдела мочевыводящих путей (4-8 неделя эмбриогенеза. Многие пороки развития являются наследственными или семейными. Кроме того, они нередко встречаются при хромосомных синдромах.
Причина облитерации мочеточника в отсутствии канализации (возникновения канала) мочеточникового тяжа. Это способны вызывать различные тератогенные факторы, воздействующие на плод в период формирования мочеточников.
К обструкции мочеточников существует семейная предрасположенность. Предполагается полигенный тип наследования. Кроме того, существует сцепленность аномалии с полом: в два раза чаще поражаются мальчики.
Стриктуры мочеточников могут сочетаться со стриктурами в желудочно-кишечном тракте, что вызывает предположения об идентичности их причины. Часто это аномалия сочетается с дисплазией соответствующей почки.
В диагностике пороков развития почек и мочевыводящих путей используются такие методы исследования, как УЗИ почек, рентгенография с контрастом. Может потребоваться ангиография (при добавочных артериях).
Пороки мочевыводящих путей без своевременного хирургического лечения приводят к нарушению оттока мочи, гидронефрозу, образованию камней, развитию хронического пиелонефрита и к почечной недостаточности. Наиболее тяжелые из них (агенезия, атрезия мочевых путей) приводят к смерти от уремии вскоре после рождения.
Читайте также: