Вторичные гемофагоцитарные лимфогистиоцитозы (ВГС) - клиника, диагностика
Добавил пользователь Alex Обновлено: 09.01.2026
Вторичные гемофагоцитарные лимфогистиоцитозы (ВГС) - клиника, диагностика
Вторичные гемофагоцитарные лимфогистиоцитозы, или, как их чаще называют, вторичные гемофагоцитарные синдромы (ВГС), — группа заболеваний, в основе которых лежат аномальная активация и пролиферация Т-лимфоцитов и макрофагов. Клинически вторичные гемофагоцитарные лимфогистиоцитозы проявляются тем же симптомокомплексом, что и первичные варианты: лихорадка, гепатоспленомегалия, панцитопения, гипокоагуляция, неврологическая симптоматика.
При постановке диагноза вторичных гемофагоцитарных лимфогистиоцитозов необходимо использовать критерии, разработанные для первичных гемофагоцитарных лимфогистиоцитозов, хотя при вторичных формах данный симптомокомплекс может быть неполным. В зависимости от природы основного заболевания, явившегося фоном для развития вторичных гемофагоцитарных лимфогистиоцитозов, принято выделять гемофа-гоцитарный синдром, ассоциированный с инфекцией (ИАГС), гемофагоцитарный синдром, ассоциированный со злокачественными опухолями (ОАГС), а также гемофагоцитарный синдром, ассоциированный с аутоиммунными заболеваниями. Нередко вторичные гемофагоцитарные лимфогистиоцитозы развивается на фоне иммуносупрессивной терапии, например, у пациентов после трансплантации органов.
В отличие от первичных гемофагоцитарных лимфогистиоцитозов, поражающего детей раннего возраста, вторичные гемофагоцитарные лимфогистиоцитозы могут развиться в различном возрасте, в том числе и у взрослых. Часто бывает трудно провести грань между вторичной и первичной формами лимфогистиоцитозов, так как вирусная инфекция, нередко являющаяся причиной развития вторичных форм, может провоцировать манифестацию наследственных форм этого заболевания.

Кроме того, если ранее считалось, что семейная форма лимфогистиоцитоза характерна исключительно для детей раннего возраста, то после частичной расшифровки генетической природы первичных гемофагоцитарных лимфогистиоцитозов стало ясно, что в некоторых случаях развитие гемофагоцитоза у взрослых связано с поздней манифестацией генетически детерминированных форм. Диагностика вторичных гемофагоцитарных лимфогистиоцитозов крайне сложна, поскольку не существует ни одного специфичного симптома, характерного только для этого заболевания.
В связи с этим при постановке диагноза вторичного гемофагоцитарного лимфогистиоцитоза врачу необходимо тщательно анализировать весь симптомокомплекс, помня о том, что если у пациента на фоне иммуносупрессии либо тяжелой вирусной инфекции остро развивается высокая рефрактерная лихорадка, гепатоспленомегалия, панцитопения и другие характерные симптомы, то диагноз лимфогистиоцитоза высоковероятен независимо от того, обнаруживаются или нет гемофагоцитирующие клетки в костном мозге. Важно подчеркнуть, что гемофагоцитарный синдром является крайне тяжелым, часто смертельным заболеванием, и только своевременная диагностика и адекватная терапия позволяют спасти пациента.
Особую группу составляют гемофагоцитарные синдромы при таких наследственных иммунодефицитах, как синдром Чедиака—Хигаси, Х-сцепленный лимфопролиферативный синдром (ХЛПС), синдром Гриссели. В редких случаях вторичный гемофагоцитарный лимфогистиоцитоз может развиться как следствие метаболических расстройств, таких, как перегрузка липидами при парентеральном питании и наследственный дефект метаболизма лизина, а также при терапии фенитоином.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Диагностика ассоциированного с инфекцией гемофагоцитарного синдрома (ИАГС) - дифференциация
Патоморфологическая картина ассоциированного с инфекцией лимфогистиоцитоза идентична таковой при семейной форме заболевания. Обнаруживается выраженная лимфогистиоцитарная инфильтрация печени, селезенки, костного мозга. Гистиоциты не несут черт гистологической атипии и активно вовлечены в фагоцитоз клеточных элементов крови и их костно-мозговых предшественников.
Как и при первичных формах, гемофагоцитоз удается выявить приблизительно у половины пациентов. Это связано с очаговостью поражения и периодом заболевания: на ранних стадиях гемофагоцитоз выявляется реже, чем в продвинутой фазе. В костном мозге массивная лимфогистиоцитарная инфильтрация закономерно сменяется интенсивным гемофагоцитозом и далее аплазией. В лимфатических узлах и селезенке отмечается атрофия зародышевых центров.
Иммунопатогенез гемофагоцитарного синдрома ассоциированного с инфекцией
Общие механизмы патогенеза как первичного, так и вторичных гемофагоцитарных синдромом идентичны. Вероятно, взаимодействие генетически полиморфных систем негативного контроля иммунного ответа, иммуносупрессивных препаратов и микробных факторов определяет клинические проявления гемофагоцитарного синдрома.
Среди экспериментально доказанных механизмов гиперактивации иммунной системы при гемофагоцитарном синдроме ассоциированном с инфекцией наиболее значимым является прямая индукция выработки ФНОа при инфицировании Т-лимфоцитов вирусом Эпштейна—Барр. Интересно, что в этой системе инкубация ЭБВ-инфицированных Т-лимфоцитов с моноцитарной клеточной линией приводила к усилению фагоцитарной активности моноцитов и продукции ИЛ-1а, ФНОа и ИФН-у, тогда как обработка анти-ФНО и анти-ИФН блокировала этот эффект.
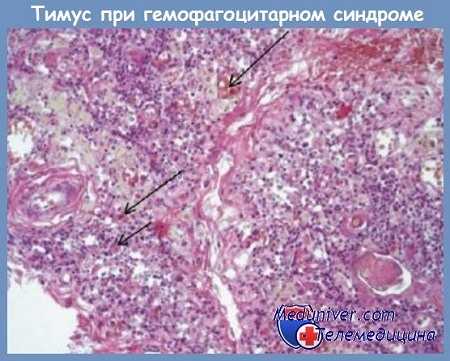
Дифференциальная диагностика гемофагоцитарного синдрома ассоциированного с инфекцией
Подход к дифференциальной диагностике гемофагоцитарного синдрома ассоциированного с инфекцией в значительной степени определяется возрастом пациентов. В педиатрической популяции центральным вопросом является выявление генетически детерминированных форм лимфогистиоцитоза. Как отмечалось, даже в случае обнаружения инфекционного агента необходимо тщательно проанализировать факторы, свидетельствующие в пользу наследственной формы заболевания.
Развитие болезни на 1-м году жизни, положительный семейный анамнез характерны для первичного гемофагоцитарного лимфогистиоцитоза. Определение экспрессии перфорина на NK-клетках с помощью проточной цитофлюориметрии и молекулярно-генетический анализ гена перфорина позволяют верифицировать диагноз приблизительно у 30 % больных с семейным лимфогистиоцитозом. Сочетание гемофагоцитарного синдрома и альбинизма встречается при двух наследственных синдромах: синдроме Чедиака—Хигаси и синдроме Гриссели.
Указание на Х-сцепленный характер наследования, т. е. развитие сходного заболевания у родственников мужского пола по материнской линии, делает вероятным диагноз Х-сцепленного лимфопролиферативного синдрома (ХЛПС, синдром Пуртило). У взрослых больных основной задачей является выявление злокачественного новообразования, так как часто гемофагоцитарный синдром может явиться единственным проявлением скрытой опухоли.
Лечение ассоциированного с инфекцией гемофагоцитарного синдрома (ИАГС)
Универсальной стандартизованной патогенетической терапии вторичных форм лимфогистиоцитозов не существует. У части больных гемофагоцитарный синдром разрешается спонтанно, и эти пациенты нуждаются только в сопроводительной терапии. Большое число пациентов умирают от неконтролируемой лимфогистиоцитарной пролиферации, несмотря на интенсивную иммуносупрессивную терапию. Серьезным психологическим барьером становится необходимость применения химиотерапии у пациентов с реактивным по своей природе «незлокачественным» заболеванием.
Среди основных направлений патогенетической терапии вторичных гемофагоцитарных синдромов можно выделить следующие:
1) терапия внутривенным иммуноглобулином;
2) плазмаферез;
3) кортикостероиды;
4) интенсивная иммуносупрессия (циклоспорин А, антитимоцитарный глобулин);
5) химиотерапия;
6) трансплантация костного мозга.
Терапия высокими дозами иммуноглобулина для внутривенного введения (1—3 г/кг на курс) является общепринятым методом при гемофагоцитарном синдроме ассоциированном с инфекцией. Такое лечение оказывает иммуномодулирующий эффект за счет блокады макрофагальных Fc-рецепторов и облегчает контроль инфекционного процесса. Плазмаферез позволяет быстро взять под контроль гиперцитокинемию, может применяться как средство «скорой помощи», однако в качестве монотерапии неэффективен в связи с явлением «рикошета».

Кортикостероиды в разных режимах остаются основой иммуносупрессивной терапии ГЛГ. Базовыми препаратами являются дексаметазон (10 мг/м2 в сутки) и метилпреднизолон (2—30 мг/кг в сутки).
Циклоспорин А (5 мг/кг в сутки) может быть эффективен как в инициальной терапии, так и для поддержания достигнутой ремиссии.
АТГ является мощным иммуносупрессивным агентом, однако чаще применяется как препарат второй линии.
Наиболее эффективной оказалась комбинация дексаметазона с этопозидом — химиопрепаратом, высокоактивным в отношении клеток макрофагального ряда.
Результаты лечения детей с семейным вариантом ГЛГ по протоколу HLH-94 позволяют рекомендовать данную комбинацию в качестве терапии первой линии.
Учитывая высокую смертность пациентов со вторичными формами лимфогистиоцитозов, курс интенсивной иммунохимиотерапии представляется оправданным и часто оказывает четко выраженный положительный эффект. Наиболее сложным является решение о продолжении терапии. Следует руководствоваться следующими принципами:
1) при отсутствии ремиссии оправдана попытка экспериментальной химиотерапии и выполнение пересадки костного мозга;
2) при появлении доказательств наследственной природы гемофагоцитарного синдрома пересадка костного мозга должна быть выполнена независимо от ответа на терапию;
3) при достижении полной ремиссии возможна отмена терапии, однако, если в процессе клинического наблюдения регистрируется рецидив гемофагоцитарного синдрома, ТКМ вновь становится единственным шансом на излечение.
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз – группа врожденных и приобретенных заболеваний, возникающих вследствие нарушений регуляции иммунного ответа и характеризующихся гиперпродукцией гистиоцитов, а также цитотоксических T-лимфоцитов. Клиническая симптоматика проявляется фебрильной лихорадкой, увеличением печени и селезенки, периферических лимфатических узлов, поражением нервной системы, костного мозга и других органов. Диагностика основана на данных клинического, лабораторного (цитопения, коагулопатия, билирубинемия и др.) и инструментального обследования. Лечение: иммуносупрессивная терапия, кортикостероиды, воздействие на причинные факторы.
МКБ-10
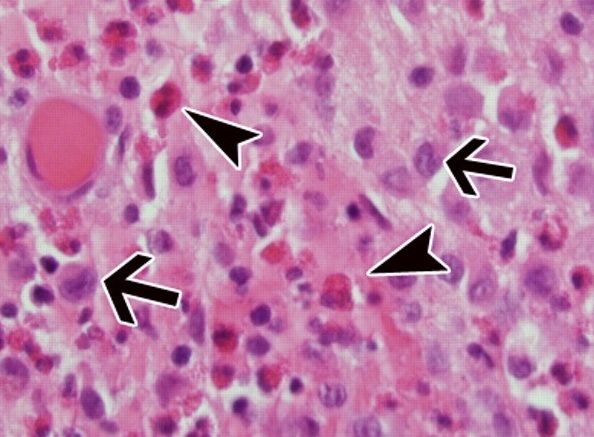
Общие сведения
Гемофагоцитарный лимфогистиоцитоз (гемофагоцитарный синдром) – врожденное или приобретенное нарушение регуляции иммунного ответа, при котором происходит аномальная активация цитотоксических T-лимфоцитов, моноцитов и макрофагов с аккумуляцией в органах-мишенях и развитием в них выраженного патологического процесса (воспаления, повреждения тканей, фагоцитоза форменных элементов крови). При врожденной, генетически детерминированной форме заболевания болеют преимущественно дети раннего возраста и в 60-80% случаев – на первом году жизни.
Вторичный (приобретенный) гемофагоцитарный лимфогистиоцитоз встречается во всех возрастных категориях, развивается на фоне затяжного течения различных инфекционных заболеваний, аутоиммунных процессов и новообразований. Впервые признаки гемофагоцитарного синдрома были описаны еще в 1939 году, а заболевание тогда было названо гистиоцитарным медуллярным ретикулезом. Семейная наследственная форма лимфогистиоцитоза была впервые описана в 1959 году. Распространенность заболевания колеблется от 1 случая на 50 тысяч новорожденных до 1-2 случаев на 1 миллион детей в возрасте до 15 лет.
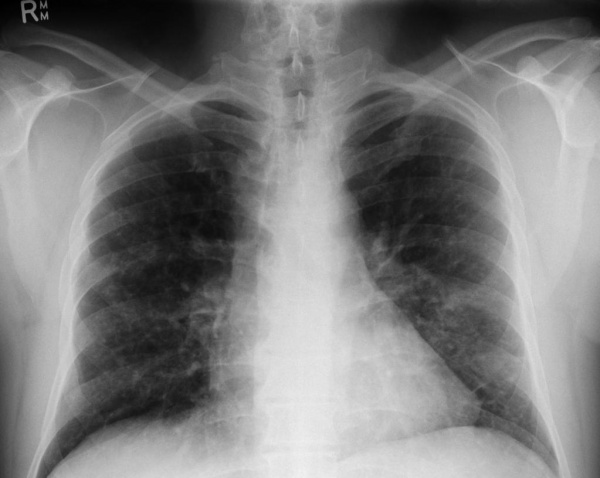
Причины гемофагоцитарного лимфогистиоцитоза
Врожденный гемофагоцитарный лимфогистиоцитоз возникает вследствие генетического дефекта механизмов клеточной цитотоксичности из-за мутаций гена перфорина. В норме регуляция иммунного ответа обеспечивается своевременным ограничением активности эффекторов иммунной системы в процессе ликвидации угрозы для организма со стороны проникших инфекционных агентов и других негативных воздействий. В этой регуляции важную роль играют механизмы клеточной цитотоксичности.
При первичном гемофагоцитарном лимфогистиоцитозе регулятивная роль цитотоксических гранул клеток и цитотоксических T-лимфоцитов в отношении клеток-мишеней нарушается, происходит чрезмерная активация иммунных клеток, в избытке продуцируются провоспалительные цитокины (интерфероны, фактор некроза опухоли и др.). Активированные вследствие «цитокинового шторма» T-лимфоциты и макрофаги инфильтрируют, а затем повреждают органы и ткани. Важным фактором патогенеза заболевания является развитие патологического гемофагоцитоза зрелых форменных элементов крови. Такой фагоцитоз происходит и в норме, помогая организму избавиться от старых клеток. В данном же случае макрофаги начинают фагоцитировать полноценные, нормально функционирующие форменные элементы крови, приводя к выраженной цитопении, коагулопатии и другим проявлениям заболевания.
Приобретенный (вторичный) гемофагоцитарный лимфогистиоцитоз развивается на фоне некоторых инфекционных заболеваний, опухолей, аутоиммунных процессов, при трансплантации органов и тканей, причем характерные нарушения регуляции иммунного ответа могут быть как следствием основного заболевания, так и осложнением, связанным с проведением иммуносупрессивной терапии и возникновением вторичной инфекции.
Симптомы гемофагоцитарного лимфогистиоцитоза
Клинические проявления гемофагоцитарного лимфогистиоцитоза чрезвычайно вариабельны. Наиболее часто наблюдается длительная лихорадка, рефрактерная к проводимой антибактериальной и противовирусной терапии. Озноб, признаки общей интоксикации (слабость, потливость, нарушения сна, отказ от приема пищи, тошнота и рвота, боли в мышцах) продолжаются в течение длительного времени, приобретая волнообразное течение с периодическими временными улучшениями самочувствия.
Характерным проявлением заболевания является увеличение печени и селезенки, имеющее прогрессирующий характер. К ранним симптомам первичного гемофагоцитарного лимфогистиоцитоза относится появление кожной сыпи, увеличение периферических лимфатических узлов, а также развитие неврологических расстройств в виде повышенной возбудимости, двигательных нарушений и расстройств чувствительности, судорожного синдрома, признаков повышения внутричерепного давления у детей раннего возраста. Встречаются при гемофагоцитарном лимфогистиоцитозе и симптомы, свидетельствующие о наличии анемии, коагулопатии – бледность и желтушность кожных покровов, периферические отеки, признаки кровотечения из пищеварительного тракта (черный кал, примесь крови в каловых массах и др.).
При вторичном гемофагоцитарном синдроме сочетаются клинические признаки поражения иммунной системы и проявления основного заболевания (вирусной инфекции, злокачественного новообразования, аутоиммунного заболевания).
Диагностика гемофагоцитарного лимфогистиоцитоза
Диагноз устанавливается в результате тщательного изучения анамнеза, клинической картины заболевания, результатов лабораторных и инструментальных исследований. Необходимы осмотры врача-гематолога, аллерголога-иммунолога, онколога, инфекциониста, ревматолога и других специалистов. Разработаны международные диагностические критерии гемофагоцитарного лимфогистиоцитоза, к которым относятся:
- лихорадка с повышением температуры выше 38,5 градусов, продолжающаяся более недели;
- увеличение печени и селезенки;
- цитопения с уменьшением гемоглобина ниже 90 г/л, тромбоцитов – меньше 100000 клеток/мкл, нейтрофилов – меньше 1000/мкл;
- признаки коагулопатии;
- увеличение ферритина больше 500 нг/мл;
- повышение уровня растворимого sCD25 в крови;
- низкое или полное отсутствие активности NK-клеток.
Приобретенные формы гемофагоцитарного синдрома диагностируются на основании вышеописанных международных критериев и проведения уточненной диагностики основного заболевания, для чего зачастую необходимо выполнять сложные лабораторные и инструментальные исследования (эндоскопические, УЗИ, КТ, МРТ, ПЭТ).
Дифференциальный диагноз гемофагоцитарного лимфогистиоцитоза проводится с:
- различными врожденными и приобретенными иммунными заболеваниями,
- болезнями крови,
- острыми и хроническими вирусными инфекциями,
- злокачественными новообразованиями (острым лимфобластным лейкозом, неходжкинскими лимфомами, другими злокачественными опухолями после проведенной химиотерапии),
- системными заболеваниями соединительной ткани: системной красной волчанкой, ювенильным дерматомиозитом, узелковым периартериитом, ювенильным ревматоидным артритом.
Лечение гемофагоцитарного лимфогистиоцитоза
Современная тактика лечения наследственной формы гемофагоцитарного лимфогистиоцитоза включает проведение химиотерапии с использованием иммуносупрессивных средств (дексаметазона, этопозида, циклоспорина A), а также трансплантации стволовых клеток. Прогноз заболевания значительно улучшается при своевременном проведении трансплантации от гистосовместимого родственного донора.
При вирусных, бактериальных и паразитарных инфекциях, вызвавших появление гемофагоцитарного синдрома, проводится этиотропная антимикробная терапия, инфузии высокодозного иммуноглобулина, а также иммуносупрессивная терапия с введением циклоспорина A и кортикостероидов. Иногда показано проведение трансплантации костного мозга. При опухолях, наряду с лечением основного заболевания, в терапевтическую схему включаются иммуносупрессивные средства в индивидуально подобранных дозах. При аутоиммунных процессах лечение включает сочетание иммуноглобулина, пульс-терапии с использованием кортикостероидов, а также циклоспорина A.
Гистиоцитоз X ( Гистиоцитоз из клеток Лангерганса , Легочная эозинофильная гранулема )
Гистиоцитоз Х – это системное заболевание, характеризующееся образованием специфических клеточных гранулем в различных органах и тканях. Наиболее типичные клинические проявления включают кожные высыпания, кашель, односторонний экзофтальм. Также наблюдаются выпадение зубов, увеличение периферических лимфатических узлов, признаки несахарного диабета (полиурия, полидипсия). Диагноз верифицируется путем гистологического исследования кожи, легких или лимфатических узлов. В качестве лечения применяются противовоспалительные препараты (глюкокортикостероиды), химиотерапевтические средства. В случае выраженного поражения легких производится трансплантация органа.
Гистиоцитоз Х (гистиоцитоз из клеток Лангерганса, легочная эозинофильная гранулема) – системная патология, при которой происходит активная пролиферация клеток Лангерганса в тканях легких, костей, центральной нервной системы, ретикуло-эндотелиальной системы (в печени, селезенке, лимфатических узлах). Течение заболевания варьирует в зависимости от формы и может быть доброкачественным со спонтанной ремиссией или быстропрогрессирующим с высокой вероятностью летального исхода. Распространенность гистиоцитоза составляет 5 на 1 000 000 человек. Преимущественно страдают дети, подростки, взрослые в возрасте 20-30 лет. Гистиоцитоз встречается только у представителей белой расы, чаще у мужчин (соотношение с женщинами 2:1).
Причины
На сегодняшний день точная причина гистиоцитоза неизвестна. Предполагается роль иммунной аутоагрессии, вызванной инфицированием вируса герпеса 6-го типа. До сих пор ведутся научные дебаты о наследственной природе заболевания. У 50% пациентов в патологических клетках Лангерганса была обнаружена соматическая мутация гена V600E, кодирующего внутриклеточный сигнальный белок BRAF. Табакокурение рассматривается как один из основных факторов риска (более 90% больных являются курильщиками). Подтверждением этого служит тот факт, что симптоматика уменьшается после прекращения курения даже без применения какого-либо лечения.
Патогенез
Механизм развития гистиоцитоза изучен недостаточно. Основным звеном патогенеза считается накопление в тканях дендритных клеток (клеток Лангерганса, или гистиоцитов). Дендритные клетки синтезируются в костном мозге и мигрируют в дерму, паренхиму легких, ретикулоэндотелиальную систему и т.д. Их основная функция заключается в поглощении антигенов, поступающих из окружающей среды, и презентации Т и В-лимфоцитам для формирования иммунного ответа. После контакта с гистиоцитами лимфоциты начинают выделять провоспалительные цитокины и медиаторы, повышающие активность гистиоцитов и придающие им высокую подвижность.
При гистиоцитозе по неизвестным причинам у дендритных клеток нарушается процесс апоптоза (запрограммированной клеточной гибели). В сочетании с выделяемыми лимфоцитами факторами роста это приводит к их интенсивной пролиферации с последующим слиянием с эозинофилами. В результате образуются гигантоклеточные гранулемы, которые постепенно начинают замещать нормально функционирующую ткань того или иного органа. Присутствие большого количества Т-лимфоцитов в гранулемах позволяет предположить наличие специфического антигена.
Классификация
По локализации очагов выделяют моносистемную форму с единичным или множественным поражением одной анатомической области и мультисистемную форму с признаками нарушения функции органов или без них. Традиционно различают следующие клинические формы:
- Диссеминированная (первично-острая, болезнь Абта—Леттерера—Сиве). Течение напоминает тяжелую системную инфекцию. Характеризуется быстрой генерализацией процесса, прогрессированием легочной недостаточности и высокой частотой летальности. Чаще возникает у детей от 6 месяцев до 2 лет.
- Первично-хроническая (болезнь Хенда—Шюллера—Крисчена). Течение также системное, но более благоприятное, различные органы вовлекаются постепенно. Типична триада Крисчена – несахарный диабет, односторонний экзофтальм и деструкция плоских костей черепа. Встречается у детей и подростков.
- Эозинофильная гранулема(болезнь Таратынова). Наиболее доброкачественный вид. Преимущественная локализация гранулем - кости и легкие. В большинстве случаев развивается у взрослых.
Симптомы гистиоцитоза
Клиническая картина крайне разнообразна. Заболевание начинается с появления общей слабости, повышения температуры тела, которая в основном бывает субфебрильной, но может достигать 40°С. Возможны диспепсические нарушения – тошнота, рвота, диарея. Кожа покрывается зудящими очагами гиперемии с чешуйками или корочками. Иногда наблюдаются геморрагические элементы, длительно незаживающие язвы. Высыпания присутствуют на коже головы, наружного слухового прохода, в области естественных складок (паховой, подмышечной). Увеличиваются лимфатические узлы по всему телу.
Возникает одышка, упорный сухой кашель и боли в грудной клетке. Затруднение дыхания может беспокоить только во время физической нагрузки или присутствовать даже в покое. Присоединяются тупые боли или тяжесть в правом подреберье из-за увеличения печени (гепатомегалии). При выраженном поражении печени кожа приобретает желтушный оттенок. Увеличение селезенки сопровождается болью в левом подреберье. Некоторые пациенты испытывают боль в костях. Деструкция костей глазницы ведет к образованию экзофтальма, чаще одностороннего. Боль в ухе ошибочно диагностируется как бактериальный или грибковый отит и безуспешно лечится антибактериальными и противогрибковыми препаратами. Остеолизис нижней челюсти приводит к периодонтитам и выпадению зубов.
При длительном течении гистиоцитоза больной начинает терять вес. В тяжелых случаях встречаются признаки костномозговой недостаточности – анемический синдром (бледность кожи, головокружение, учащенное сердцебиение), склонность к кровотечениям, сниженная сопротивляемость к инфекциям. Формирование гистиоцитарных гранулем в задней доле гипофиза вызывает угнетение выработки антидиуретического гормона. В результате развивается симптоматика несахарного диабета – сухость во рту, постоянная жажда (полидипсия) и повышенное мочеотделение (полиурия).
Осложнения
Широкая вариативность клинической картины при гистиоцитозе обусловливает разнообразие осложнений. Наиболее характерными считаются патологические переломы костей, особенно компрессионный перелом позвоночника. Типичные осложнения со стороны легких – легочная артериальная гипертензия вследствие диффузного фиброза легочной ткани, спонтанный пневмоторакс из-за разрыва тонкостенных булл. Массивная инфильтрация печени вызывает цирроз с печеночно-клеточной недостаточностью. К редким осложнениям относятся неблагоприятные последствия дефицита антидиуретического гормона (несахарного диабета) в виде гиперосмолярной гипогидратации, проявляющейся двигательным беспокойством, мышечными судорогами, нарушением сознания, вплоть до глубокой комы.
Диагностика
Пациентами с гистиоцитозом занимаются различные врачи – гематологи, пульмонологи, педиатры. Профиль специальности зависит от возраста больного и преимущественно пораженного органа. При общем осмотре отмечаются цианоз (синюшность) губ, участие в дыхании вспомогательной мускулатуры (при дыхательной недостаточности). При аускультации легких выслушивается жесткое дыхание, сухие хрипы по всем легочным полям. Дополнительное обследование включает:
- Лабораторные тесты. В общем анализе крови обнаруживаются ускорение скорости оседания эритроцитов, иногда эозинофилия и панцитопения (уменьшение эритроцитов, тромбоцитов и лейкоцитов). В биохимии крови может прослеживаться увеличение печеночных ферментов (АЛТ, АСТ) и повышение осмолярности плазмы. При печеночной недостаточности изменяются показатели коагулограммы (удлинение протромбинового времени, гипофибриногенемия). В анализе мочи наблюдается низкая относительная плотность (гипостенурия).
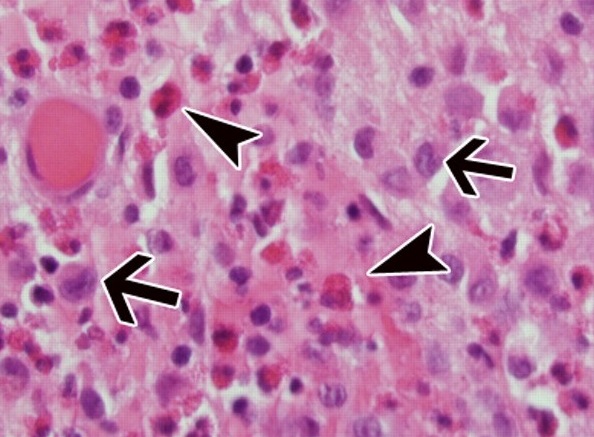
- Инструментальные исследования. При рентгенографии скелета находят очаги деструкции и остеолизиса, особенно выраженные в плоских костях черепа и длинных трубчатых костях. На рентгенограмме органов грудной клетки видны двусторонние мелкоочаговые затемнения (узелки, буллы), усиление легочного рисунка. Для более точной визуализации легочной ткани назначается компьютерная томография легких высокого разрешения, позволяющая определить их ячеистую деформацию. Результаты измерения функции внешнего дыхания (спирографии) показывают ухудшение диффузионной емкости легких.
- Верифицирующие тесты. Позволяют достоверно установить диагноз гистиоцитоза. При гистологическом исследовании биоптата (чаще кожи, лимфатических узлов или легких) выявляется избыточное количество гигантских клеток Лангерганса с эозинофильной цитоплазмой, бобовидной формой ядра, отсутствием ядрышек. Для идентификации поверхностных специфических маркеров гистиоцитоза (CD 1а и лангерина) проводится иммуногистохимический анализ.
Дифференциальный ряд включает большое количество нозологий и зависит от клинических симптомов гистиоцитоза. Кожные высыпания нужно дифференцировать с экземой, псориазом, атопическим дерматитом. Поражение легких следует отличать от туберкулеза, саркоидоза, лимфогранулематоза. Генерализованный гистиоцитоз дифференцируют с гематологическими заболеваниями (гемофагоцитарный синдром, острый лейкоз). Очаги деструкции костей требуют исключения гиперпаратиреоза, множественной миеломы, остеомиелита.
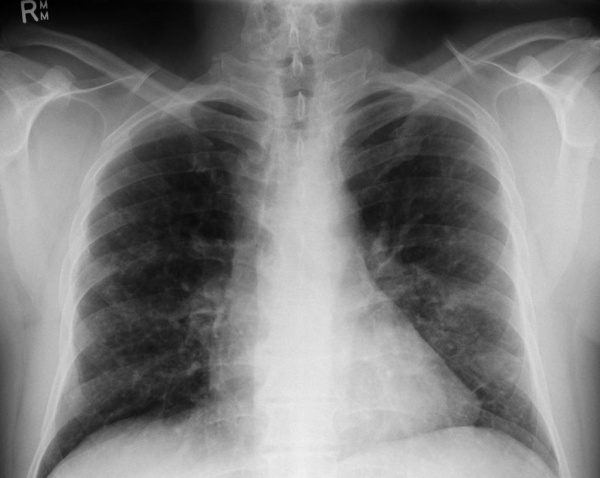
Рентгенография легких. Диффузные двусторонние узловые затемнения и небольшие кисты в верхней и средней доле
Лечение гистиоцитоза
Пациенты обязательно должны быть госпитализированы в стационар. При тяжелой дыхательной недостаточности показаны ингаляции кислорода или перевод на искусственную вентиляцию легких. При наличии признаков костномозговой недостаточности прибегают к переливанию компонентов крови и применению гранулоцитарного колониестимулирующего фактора.
Этиотропной терапии гистиоцитоза Х не существует. Наиболее важный этап в лечении – отказ от курения. Прекращение курения вызывает улучшение клинической, лабораторной, рентгенологической картины. В качестве патогенетической терапии используют глюкокортикостероиды (преднизолон, метилпреднизолон), химиотерапевтические средства (винбластин, меркаптопурин, этопозид).
Для лечения несахарного диабета назначается заместительная гормональная терапия аналогами вазопрессина (десмопрессин) в виде интраназального спрея или в таблетированной форме. При небольших остеолитических очагах выполняют кюретаж, при выраженной инфильтрации костей – резекцию или дистанционную гамма-терапию. При массивном деструктивном процессе в легочной ткани проводится трансплантация легких.
Прогноз и профилактика
Течение и прогноз при гистиоцитозе Х определяется формой заболевания. Первично-острая форма характеризуется быстропрогрессирующим течением с неблагоприятным прогнозом. Смерть наступает в 70-80% случаев от легочно-сердечной недостаточности. Первично-хроническая форма и эозинофильная гранулема имеют более доброкачественное течение. Риск летальности составляет 15% и 1,5% соответственно. Иногда происходит спонтанное выздоровление. Так как причина развития гистиоцитоза неизвестна, эффективных методов профилактики не разработано. Уменьшить риск возникновения и рецидива может отказ от табакокурения.
1. Доброкачественное течение гистиоцитоза Х/ Шихнебиев Д.А., Эседов Э.М., Джалилова Л.М.// Клиническая медицина. – 2002 - №7.
2. Роль дерматолога в диагностике гистиоцитоза из клеток Лангерганса/ Мошкалова И.А.// Учебные записки СПбГМУ им. акад. И.П. Павлова. - 2001, т. VIII, №4.
4. Легочный гистиоцитоз Х — современные представления, диагностика и тактика лечения/ Корнев Б.М., Коган Е.А., Попов Е.Н. и др.// Терапевтический архив - 2003 - №5.
Читайте также:
- Кашлевый рефлекс. Дыхательные функции носа
- Врожденные пороки половых органов
- Клиника гнойного менингита. Неврологические симптомы гнойного менингита. Тактика при гнойном менингите.
- Детренированность при длительном действии невесомости. Глубоководные погружения
- КТ, МРТ при хордоме паравертебрального пространства