Ювенильная ксантогранулема у новорожденного
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Заболевание встречается у детей раннего возраста, в основном до 24 месяцев.Почти в 75% случаев высыпания появляются в течении первого года, а в 15% случаев они наблюдаются при рождении.
Описаны случаи появления ксантогранулем у взрослых, как правило в 20-30 лет, но могут встречаться и у пожилых.
При одиночных высыпаниях заболевание несколько чаще появляется у мальчиков, чем у девочек в соотношении 1, 5:1, но при множественных поражениях преобладание среди лиц мужского пола, по сравнении с женским, резко возрастает и составляет 12:1.Среди детей европеоидной расы заболевание встречается в 10 раз чаще, чем у других рас.
Патогенез заболевания неясен.Заболевание является доброкачественной опухолью из дифференцированных гистиоцитов не- лангергансового типа. Уровень липидов в сыворотке крови нормальный. Причина прогрессивной липидизации (накопления липидов) гистиоцитов при отсутствии гиперлипидемии неясна.Однако было показано, что у взрослых пациентов с ксантогранулемой увеличивается поглощение липопротеинов низкой плотности и синтез холестерина в макрофагах.Есть мнение, что гистиоциты, возможно, реагируют таким образом на травматический или инфекционный факторы.
Некоторые авторы предполагают, что что генерализованная эруптивная гистиоцитома, доброкачественный цефальный гистиоцитоз и ювенильная ксантогранулема могут представлять собой различные проявления одного и того же заболевания.Ювенильная ксантогранулема не является наследственным заболеванием, но в ряде случаев ассоциируется с нейрофиброматозом I типа (NF1) и ювенильной миеломоноцитарной лейкемией.У некоторых пациентов наблюдается «тройная ассоциация» ксантогранулема+нейрофиброматоз+ ювенильный миеломоноцитарный лейкоз.Сообщалось также о связи ювенильной ксантогранулемой у взрослых с множественными поражениями с хронической лимфоцитарной лейкемией и В-клеточной лимфомой.
Заболевание начинается с появления розовых, красновато-серых пятен, которые быстро трансформируются в папулы, бляшки и узлы, умеренно плотные и эластичные при пальпации, с четкими границами, часто с периферическим розовым венчиком.Вначале заболевания цвет высыпаний колеблется от розового до красно-коричневого, затем они становятся светло-желтого, оранжевого, коричневого цветов с разными оттенками.
Поверхность элементов обычно гладкая, иногда с выраженным кожным рисунком, у крупных образований на ней могут наблюдаться телангиоэктазии, а в отдельных случаях эрозии и изъязвления.Наиболее частая локализация сыпи - волосистая часть головы, лицо, шея, верхняя часть груди, реже верхние и нижние конечности, половые органы.Необычными являются элементы сыпи с гиперкератозом, подкожные и сгруппированные в бляшки.
У части больных могут наблюдаться пятна "кофе с молоком", что может свидетельствовать о связи заболевания с нейрофиброматозом.
Течение заболевание хроническое, обычно через 1-6 лет (в среднем через 2-3 года) происходит спонтанный регресс высыпаний.Инволюция элементов начинается с центрального западения и небольшого размягчения, позже они сморщиваются и исчезают, оставляя после себя гипо- и (или) гиперпигментные пятна, невыраженную атрофия или анетодермию.
Микроузелковая форма
| Характеризуется высыпанием мелких куполообразных папул диаметром 2-5 мм, расположенных рассеянно.В большинстве случаев наблюдаются множественные высыпания (иногда до сотен).Чаще выявляется у детей.Описаны диссеминированные формы. |
Крупноузловая форма
| Самый распространенный вариант ювенильной ксантогранулемы.Встречается в любом возрасте.Проявляется в виде высыпания одного или нескольких узлов диаметром 1-2 см.У взрослых чаще всего наблюдается одиночный узел. |
Бляшковидная форма
| Характеризуется высыпанием одиночной или нескольких плоских бляшек желтого или оранжевого цвета, часто с неправильными границами, иногда достигающих больших размеров.Сообщается о случаях симметричной локализации. |
Гигантская форма
| Редкая форма заболевания, как правило, наблюдаемая у девочек в возрасте до 14 месяцев, которая представлена узлом более 2 см, располагающимся обычно в области верхней части спины, который часто неправильно диагностируют как детскую гемангиому. |
Язвенная форма
| Изъязвление центральной части поверхности ксантогранулемы с ее западением, придающее элементу кратерообразный вид - редкое осложнение крупноузловой или гигантской формы. |
Оральная форма
| Характеризуется одиночным узлом красно-желтого цвета, чаще на боковой поверхности языка или на средней линии твердого неба. |
Глазная форма
| Чаще встречается в возрасте до 2 лет в 10% случаев заболевания.Чаще это односторонний процесс.Локализуется на конъюнктиве глаза с захватом лимба или без.Сочетание глазной и кожной формы ксантогранулемы по данным разных авторов встречается у 0, 5 - 1% при одиночных высыпаниях и до 40% при множественных.Обычные симптомы - эритема конъюнктивы, светобоязнь, изменение окраски радужки и увеличение глазного яблока.Серьезными осложнениями являются гифема (кровоизлияние в переднюю камеру) и глаукома, которые могут привести в слепоте. |
Системная форма
Встречается приблизительно в 4% от всех случаев заболевания и у 50% больных сочетается с кожными проявлениями.Характеризуется образованием гранулем в мышцах, печени, селезенке, легких.Реже в процесс вовлекаются яички, кости и центральная нервная система, при поражении которой наблюдается атаксия, повышенное внутричерепное давление, субдуральные кровоизлияния, задержка развития, несахарный диабет и другие неврологические нарушения.Сообщалось о смертельных исходах в случаях прогрессирующей ювенильной ксантогранулемы у новорожденных при поражении печени.
Диагноз ставится на основании клинической картины, анамнеза и гистологического исследования при котором обнаруживаются:
- В ранней стадии - скопления мономорфных гистиоцитов с обильной эозинофильной цитоплазмой, макрофаги, лимфоидные клетки и эозинофильные гранулоциты.
- В стадии развития - скопления липидов в цитоплазме гистиоцитов, придающих им пенистый «ксантоматозный» вид, гигантские клетки Тутона, клетки с размещенными ядрами в виде венчика, что является типичным для ювениальной ксантагрануломы.В инфильтрате присутствуют лимфоциты, эозинофильные гранулоциты и плазматические клетки.
- В стадии регресса наблюдается пролиферация фибробластов и фиброз.
В отличие от гистиоцитоза X у гистиоцитов нет тенденции проникать в эпидермис и при электронной микроскопии гранулы Лангерганса не выявляются.
Дети в возрасте до 2-х лет с множественными высыпаниями должны находится под наблюдением офтальмолога, а с нейрофиброматозом - гематолога.
- Красно-коричневые папулы, кожа над которыми изменена по типу апельсиновой корки
- При потирании очаги краснеют и становятся более выпуклыми, превращаются в волдыри (симптом Дарье)
- Папулы ярко-красного цвета
- Очаги не накапливают липиды, поэтому не желтеют
- При диаскопии (осмотре очага при надавливании предметным стеклом) могут менять окраску на коричневую
- Обычно коричневый или цвета загара
- Может сопровождаться гипертрихозом
Пациенты с кожной формой заболевания, ввиду самопроизвольного разрешения процесса, лечению не подлежат.По косметическим причинам возможно хирургическое иссечение элементов.
При глазной форме заболевания рекомендованы кортикостероиды в виде глазных капель, субконъюнктивальных инъекция, перорального применения.Для лечения гифемы или глаукомы применяется хирургическое вмешательство, химиотерапия или лучевая терапия с низкой «катарактогенной» дозой (300-400 кГр).
При системной форме применяется химиотерапия (цитарабин, винкристин), метотрексат, циклоспорин и преднизолон в высоких дозах.При поражениях ЦНС сообщалось об успешном применении кладрибина.
Просмотр полной версии : Ювенильная ксантогранулёма или. Ребенку 2 года.
Здравствуйте! Очень прошу помочь определить, что с нами. Ребенку 2г 2мес. Уже полтора месяца нашей истории, что делать не знаю. Написала много, т.к. не знаю, что важно. В начале января на спине выше лопаток появился как-бы прыщик или жировик бледно-розового цвета с тонкой кожей сверху размером около 1мм. ( Должна сделать отступление- на Новый год позволила конфеты, т.к. есть старший ребенок, их не спрячешь… т.е. в одно время с появлением этого "прыщика" была небольшая аллергия на внешней стороне предплечий, которая после отмены сладкого почти проходила, оставалось небольшое шелушение без покраснения. Вчера дала дольку шоколада и покраснение и шершавость снова проявились. Начала фенистил. Вместе с этим также в начале января начались запоры. Стул раз в три дня. Раньше стул был регулярно, максимум 2дня перерыв. За два месяца до НГ поставили прививку Приорикс Ребенок родился на 35 неделе, была врожденная пневмония, поэтому с прививками запаздываем.
Развитие соответствует возрасту.)
Итак, наблюдала 5дней, пробовала мазать зеленкоц, в надежде, что подсохнет. Потом имела глупость(знаю, так делать нельзя) немного соскрести стерильной иголочкой верхний слой кожи, чтобы "прорвало". Но, под кожей не было гноя, как я ожидала, а какая-то плотная консистенция. Я сразу обработала зеленкой и на следующий день мы отправились к хирургу в поликлинику. После осмотра был ответ " закупорка сальной железы, мажьте зеленкой, подсушивайте". Неделю мажем, результата нет. Идем в поликлинику к дерматологу. Мнения два-моллюск или атерома. Рекомендации-подсушивать салициловой кислотой. Две недели"сушим", появляемся на контрольный осмотр. Заметила, что не только не подсушивается, но и растет. Примерно на 1-1,5 мм за это время. Идем в клинику к другому дерматологу, там посмотрели дермоскопом ( кажется, так назвается). Диагноз - ювенильная ксантогранулема. Сделали УЗИ (овальное гипоэхогеннное образование с четкими ровными контурами, однородной внутренней структуры, размер 3,5*2,5 мм без признаков кровотока при ЦДК. Подкожно-жировой и мышечный слой в описанной области без особенностей). ОАК в норме.
На данный момент (т.е. прошло полтора месяца) размер 3,5 мм, высота 2,5мм. Консистенция стала более плотной, цвет от бледно-розового изменился к желтоватому , при нажатии бледнеет.Есть бледно-розовый ободок по контуру. И меня смущает розовая точечка в центре и то, что примерно раз-два в неделю шелушится (есть на фото), затем поверхность снова становится гладкой и блестящей. Ребенка никак не беспокоит, только нас с папой очень сильно.
Вопросы:
1. На что всё-таки это похоже?
2. Нормально ли то, что есть шелушение и розовая точечка?
3. Есть ли связь с аллергией и запорами?
4. Рост спровоцирован моим воздействием или так должно быть? Или, может, трением об одежду - как раз горловина проходит рядом с этим участком? Может, пластырем заклеивать?
5. И что с этим делать?
Очень жду ответа!
Ювенильная ксантогранулема у новорожденного
Ксантогранулема ювенильная - Juvenile xanthogranulomas ( JXGs )
Ювенильная ксантогранулема являются доброкачественным образованием; обычно протекает бессимптомно; возможно самовосстановления, красные, желтые или коричневые папулы и узелки которые приимущественно состоят из гистиоцитарных клеток, образуются в младенчестве и детстве.
Папулы или узелки могут образовываться в коже,ткани глаза и внутренних органах. Ювенильная ксантогранулема является наиболее распространенной формой гистиоцитозом не-Лангергансовских клеток.
Adamson впервые сообщил о ювенильной ксантогранулеме в английской литературе в 1905 году. Он представил ребенка, у которого развились многочисленные желто-белые папулы на теле в первые 2 недели жизни. Он назвал это множественные врожденные ксантомы. В 1912 году McDonagh представил первый обзор случаев и переименовал в состоянии nevoxanthoendothelioma (хотя состояние не связано с невусом или эндотелиальными клетками). В 1954 году Helwig и Hackney снова переименовал в ювенильная ксантогранулема , что отражает его гистопатологическое строение. Laurb a и Lain в 1937 году впервые сообщили о развитиии ювенильной ксантогранулемы во внутренних органах. Blank и др. впервые описал поражение глаз в 1949 году.
Этиология Этиология ювенильной ксантогранулемы полностью не известны. Папулы и узелки ювенильной ксантогранулемы гистологически представлены дифференцированными не-Лангергансовыми клетками гистиоцитов. По общему мнению, что клетки имеют происхождения из кожных дендроцитов. Как предполагается, ювенильная ксантогранулема , возможно, является гранулематозной реакцией гистиоцитов к неопознанному стимулу, вероятно физической или инфекционной этиологии. Данные Kraus и др., предпологает возможное происхождение из CD4 + плазмацитоидные моноцитов. Ингибирование клеточного апоптоза-видимому, играет незначительную роль при росте ксантогранулемы. Появление гигантских клеток и пенящийся содержащие липиды гистиоцитов в общем происходит поздно и очевидно -является вторичным, возможно в ответ на выработку цитокинов гистиоцитами. Уровни сывароточных липидов в крови остается нормальны на всем протяжении заболевания.
Эпидемиология Ювенильная ксантогранулема появляется у белокожих примерно в 10 раз чаще, чем у афроамериканцев. В детстве, ювенильная ксантогранулема встречается преимущественно у мальчиков (1,4:1). У взрослух заболеваемость приблизительно равна. Множественные поражения кожных возникают преимущественно у мужчин (12:01).
Возраст Около 35% случаем ювенильной ксантогранулемы проявляется при рождении, 71% случаев заболевания приходится на первый год жизни. Средний возраст при постановке диагноза составляет 22 месяцев. В большинстве случаев ювенильная ксантогранулема саморазрешается к 5 летнему возрасту. Несмотря на термин ювенильная ( младенческая) в название болезни, в 10% случаев проявляются в зрелом возрасте.
Клинически Поражение, как правило, протекает бессимптомно, имея гладкие, округлые, желтого, красного, коричневого цвета единичную папулу или множественные папулы. Наиболее частым местом появления - на голове и шее, а затем на туловище и верхних конечностях, однако ксантогранулема может возникать на любом участке кожи. До 81% случаев кожной ювенильной ксантогранулемы проявляются в виде одиночного поражения. Эта форма также наиболее распространена и при взрослой ксантогранулемы.
Были описаны две формы ювенильной ксантогранулемы:
Папулезная форма имеет размеры от 2 - до 5-мм, гладкие, твердые папулы, которые изначально красно-бурые, а затем быстро изменится на желтый. Реже узловатая форма, является круглой, от 0,5 до 2 см, просвечивающие, красно-желтого, эластичный узелки с телеангиэктазии (узелки со временем могут измениться до желто-коричневого цвета). Гигантские ювенильные ксантогранулемы относится к узловатым и могут быть больше чем 2 см (самая большая, описанных в литературе, была 10 х 5 см). Более редкие варианты включают смешанную форму характеризуется как папулезные и узловые, в которых сливаются сгруппированые папулы , и подкожная форма (примерно 5%), с единственным глубоким узлом или множественными элементами. Внекожных ювенильной ксантогранулемы встречаются редко (3,9%) и чаще всего проявляются в глазах (<1%) и периорбитальной области. Глазные ксантогранулемы обычно проявляет в радужной оболочке. Следующие по частоте встречаются в легких и печени. Реже, поражаются надпочечники, аппендикс, костей, костного мозга, центральной нервной системы , половых желез, почек, гортани, миокард, перикард, забрюшинного пространства, тонкого и толстого кишечника и селезенки.
Только 50% системных поражений сопровождается кожным проявлениями ювенильной ксантогранулемы, и эти кожные повреждения, как правило, бывают множественными, а не одиночными, папулами или узлами. Размер кожного поражения не коррелирует с наличием или отсутствием системных ксантогранулем. "Кофе с молоком" пятена наблюдается примерно в 20% у больных с папулезной формой ювенильной ксантогранулемы.
Причины Сосуществование кофе с молоком пятен и несовершеннолетних Ксантогранулема (JXG) был связан с эпилепсией. Ниман-Пика болезнь была связана с JXG. Крапивница пигментная была связана с JXG. Нейрофиброматоз типа 1 (УХЛ1) была связана с JXG. ретроспективного исследования подсчитали, что больше, чем 1 из 5 детей с NF1 в возрасте до 3-х лет будет развиваться JXG. Ювенильный хронический миелолейкоз, в настоящее время в основном упоминается как ювенильный миеломоноцитарный лейкоз (JMML), было отмечено в связи с несколькими JXGs, [13, 14, 16] и распространенность особенно высока у пациентов с сопутствующей нейрофиброматоз. Статистические данные относительно этой тройной ассоциации являются спорными; оценки показывают, что пациенты с NF1 и JXG имеют 20 - до 32-раз повышенный риск развития JMML чем пациенты с NF1 одиночку. Пациенты также были диагностированы с JMML и JXG, но без УХЛ1.
Дифференциальный диагноз
- Фиброма кожи
- Гистиоцитоз Лангерганса
- Мастоцитоз
- Шпиц невус
- Ксантомами
Биопсия кожи может быть выполнена, как для диагностики и косметический. Образец обычно состоит из полного удаления папул или узелков.
Гистологические Гистологическое исследование несовершеннолетних Ксантогранулема (JXG) демонстрирует различные результаты. Зависящих от времени прогрессии существует в развитии характерных гистологические характеристики JXG, что коррелирует с возрастом поражения. Ранние образцы биопсии выявить плотные мономорфной гистиоцитарный инфильтрата в дерме. Продолжение в подкожную клетчатку, фасции, мышцы и происходит приблизительно в одной трети случаев. [18] Старые поражения содержат пенистые клетки, Touton гигантские клетки, и иностранные гигантские клетки тела. Смешанный клеточного инфильтрата нейтрофилов, лимфоцитов, эозинофилов и (редко) тучные клетки могут быть отмечены. Старые очаги фиброза продемонстрировать. Нет гистологических различий не сообщается между кожных и системных JXG. Из-за трудности в диагностике JXG и из-за переходных присутствии Touton гигантские клетки в JXG поражений, эти классические элементы, которые не могут присутствовать в каждом конкретном случае. Гистиоциты содержат плеоморфные ядер с низким содержанием или отсутствуют фигуры митоза, и нерегулярные плотных тел. Кластерированные форме запятой органов иногда наблюдаются на электронную микроскопию, но не являются специфическими. Использование специальных красителей Важно дифференцировать от JXG Лангерганса и не histiocytoses Лангерганса клетки. [19] В JXG, гистиоциты положительны для антител против фактора XIIIa, HAM56, HHF35, KP1 (CD68), Ги M1P и Виментин и , как правило, отрицательно к CD1a и S-100. Новые отчеты также продемонстрировали CD4-положительных, который был использован в качестве доказательства того, что plasmacytic моноцитов может быть нормального клеточного типа из основных составляющих JXG, а кожный dendrocyte.
Медицинская помощь Досрочная помощь уместно в случаях ограниченного и доброкачественного характера Ювенильная ксантогранулема (JXGs). Глазных и системных поражений могут отвечать на стероиды или лучевой терапии. Редкие случаи тяжелых системных JXG потребовали одно-или мультиагентный режимов химиотерапии.
Хирургической помощи Повреждения могут быть изъяты для диагностики и косметических целях. Глазные и системные поражения иссечение обычно приводит к излечению. 2008 случай докладе описывается новорожденных с множественными кожными и печеночных Ювенильная ксантогранулема (JXGs), требующие трансплантации печени с холестазом вторичных и портальной гипертензии.
Лекарства: общая информация Системные стероиды могут быть использованы для проблемной висцерального Ювенильная ксантогранулема (JXGs). Кортикостероиды уменьшают размер висцеральных узелков. Эти вещества обладают противовоспалительными свойствами и вызывают глубокие и разнообразный метаболических эффектов. Они модифицируют иммунный ответ организма на различные раздражители. Преднизолон (Deltasone, Meticorten, Orasone) DOC для висцерального поражения. Может снизить воспаления в обратном повышенной проницаемости капилляров и подавления активности ПМН.
Дальнейшее амбулаторное Последующие визиты могут быть запланированы на регулярной основе для уверенности и контроля связанных с ним осложнений. Для пациентов с ювенильным xanthogranulomas (JXGs) и нейрофиброматоз типа 1 (УХЛ1), врачи должны следить за признаками и симптомами Ювенильный миеломоноцитарный лейкоз (JMML). [22] Пациенты с поражением глаз должны регулярно проверяться у офтальмолога для предотвращения редких осложнений, таких как глаукома.
Осложнения Осложнения, связанные с Ювенильная ксантогранулема (JXGs) редки и зависят от места и участия соответствующих условий. Вовлечение глаз может прогрессировать до глазных кровоизлияния, глаукомы или отслоения сетчатки. Эти осложнения лучшие предотвратить с помощью раннего обнаружения. Поражение ЦНС является очень редким осложнением. Печеночная недостаточность является редким, но потенциально смертельным, осложнением системной JXG.
Прогноз При отсутствии терапевтического вмешательства, ювенильный xanthogranulomas (JXGs) выравниваться с течением времени. Как кожного поражения и внекожных Эвольвентные спонтанно в течение 3-6 лет. Гиперпигментация, атрофия мягких или anetoderma может сохраняться. Повреждения могут повториться после резекции. Частота рецидивов составляет около 7%. В отсутствие нейрофиброматоз, никакие системные последствия для здоровья не вовлечены, с редкими исключениями. Бдительно экране пациентов с нейрофиброматоз и JXG для лейкемии. Глазных, неврологических, и печени редки, но могут иметь серьезные долгосрочные последствия.
Обучение пациентов Убедите пациентов и их семей. Поручить пациентов, касающиеся ассоциаций, связанных с клинической ситуации (нейрофиброматоз, глазные выводы в диффузных JXG, JMML), и прямое обучение пациентов к этим условиям.
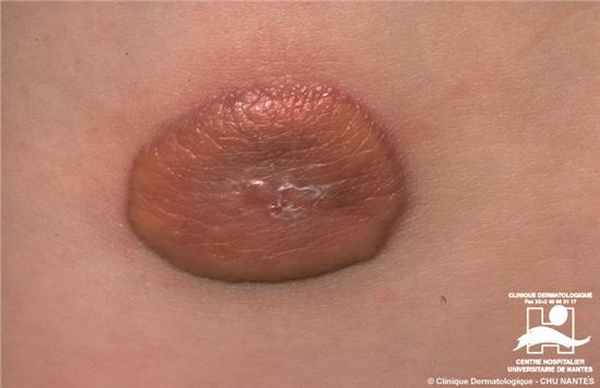
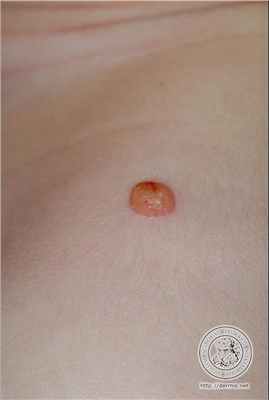
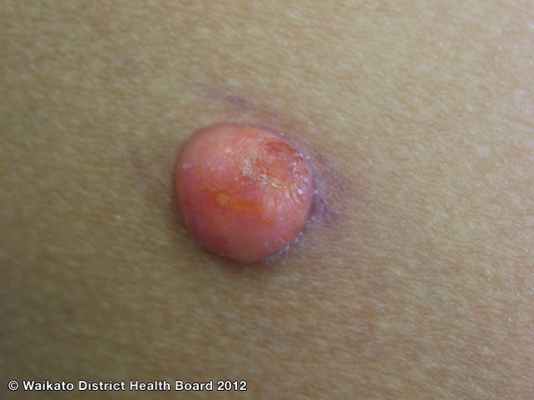
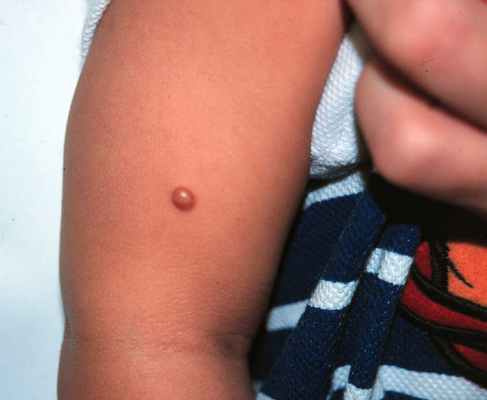
Дерматоскопия ксантогранулемы
Дерматоскопическое анализ показывает наличие периферийного кольца, окружающего центральню пигментированную область цветовая характеристикая которого колеблется от розового до желтого-оранжевого. А так же наличии точечных сосудов в центральной области.
Дерматоскопия модель: По периферии темное кольцо, центральной оранжево-желтый цвет и точечные сосуды в нем.
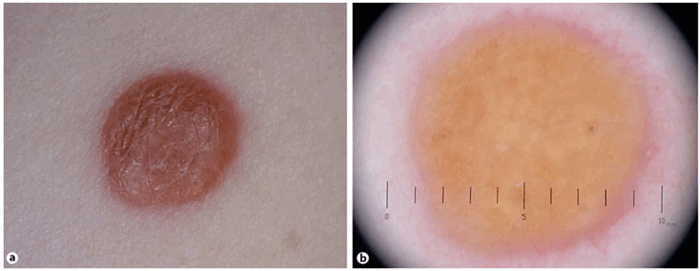
Дерматоскопия: ювенильная ксантогранулема
Улучшая цифровая цветокоррекция изображения улучшает визуализацию сосудистых структур в образовании (стрелки- рис.4)
Гистология ювенальной ксантогранулемы
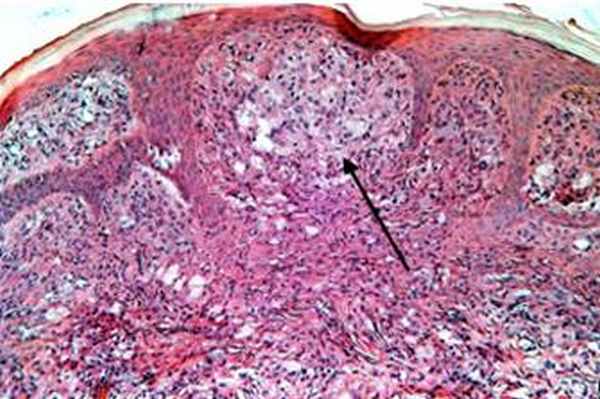
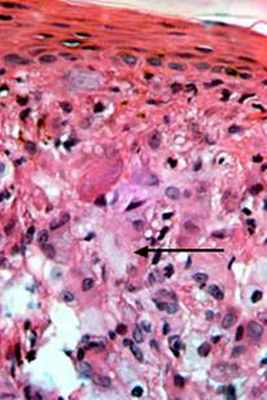
© Dermatooncology | 2022 | Dr. Alferov | Вся информация носит информационных характер в профессиональной деятельности специалиста.
Сайт содержит медицинскую информацию, выдержку печатных статей и content находящиеся в общедоступных и специализированных источниках на правах свободного доступа.
Ювенильная ксантогранулема у новорожденного
Синоним: невоксантоэндотелиома (неправильное название).
Эпидемиология ювенильной ксантогранулемы
Возраст: новорожденные (15% случаев), в возрасте до года (75% случаев).
Пол: мальчики болеют в 1,5 раза чаще.
Заболеваемость: самая распространенная форма гистиоцитоза.
Этиология: развитие реактивной гранулемы вызвано неизвестно причиной.
Патофизиология ювенильной ксантогранулемы. Реактивная пролиферация гистиоцитов в ответ на не определенный травматический или инфекционный фактор.
Анамнез ювенильной ксантогранулемы. Асимптомная кожная форма ювенильной ксантогранулемы развивается сразу или вскоре после рождения, образования могут увеличиваться в количестве и размерах в течение 1-2 дней, впоследствии как кожные, так и редкие висцеральные поражения спонтанно регрессируют в течение 3-6 лет.
Клиника ювенильной ксантогранулемы
Тип высыпаний: папулы, узлы.
Цвет: красно-коричневый, быстро переходящий в желтый.
Размер: от 2 до 20 мм в диаметре.
Количество: одиночные или множественные очаги (до нескольких сотен).
Пальпация: плотная или эластичная консистенция.
Локализация: лицо, волосистая часть головы, шея, реже - верхняя половина туловища, верхние и нижние конечности, слизистые оболочки.
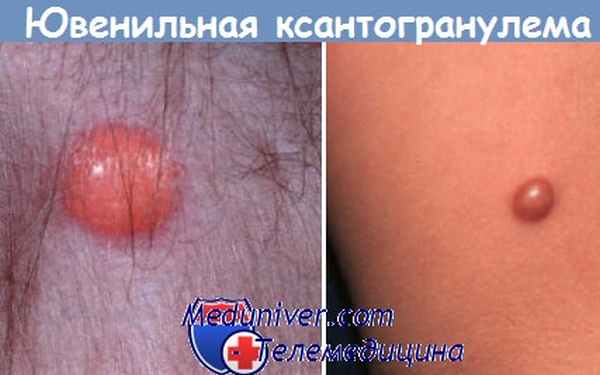
Общие проявления ювенильной ксантогранулемы
Органы зрения: глазная форма ювенильной ксантогранулемы (в 0,5% случаев).
Другие: в редких случаях отмечается сочетание с поражением легких, костей, почек, перикарда, кишечника, яичников или яичек, а также пятнами цвета «кофе с молоком» или лейкемией.
Дифференциальная диагностика ювенильной ксантогранулемы
Дифференциальный диагноз при ювенильной ксантогранулеме проводится с доброкачественным цефалическим гистиоцитозом, генерализованным эруптивным гистиоцитозом, саморазрешающимся ретикулогистиоцитозом, ксантомами, гистиоцитозом X и невусами.
Патогистология ювенильной ксантогранулемы: наблюдается мономорфный, не содержащий липидов гистиоцитарный инфильтрат, который в поверхностных слоях дермы и на периферии инфильтрата может включать пенистые клетки, гигантские клетки инородных тел и гигантские клетки Тутона. Окраска на жир положительна.
При электронной микроскопии видны гистиоциты с тельцами в форме запятой, липидными вакуолями, продукты распада холестерина и миелоидные тельца. Иммуногистохимический анализ выявляет НАМ56+, CD68+, фактор ХIIIа+.
Течение и прогноз ювенильной ксантогранулемы
Ювенильная ксантогранулема часто присутствует уже у новорожденного или развивается во время первых 9 месяцев жизни, при этом отмечается доброкачественное течение заболевания. Нередко количество высыпаний увеличивается до возраста 1-1,5 года, затем они спонтанно регрессируют. Редко поражаются глаза, легкие, перикард, мозговые оболочки, печень, селезенка, яички. Общее состояние ребенка и его развитие не нарушены.
Глазная ювенильная ксантогранулема - наиболее неблагоприятная форма заболевания, связанная с развитием таких осложнений как глаукома, кровоизлияния и слепота. В редких случаях при ювенильной ксантогранулеме определяется повышение уровня фактора некроза 1, при этом риск развития ювенильной миеломоноцитарной лейкемии значительно повышается.
Лечение ювенильной ксантогранулемы
Лечение при кожной форме заболевания не проводится, так как высыпания регрессируют самостоятельно.
При поражении глаз для предотвращения развития глаукомы, кровоизлияний и слепоты могут назначаться лучевое лечение и/или местная или системная терапия стероидами.
В редких случаях ювенильная ксантогранулема при системной форме заболевания также может спонтанно регрессировать, однако в тяжелых случаях назначается системная химиотерапия, лучевое лечение, кортикостероиды или циклоспорин.
Ювенильная ксантогранулема у новорожденного
Нейрофиброматоз 1 типа – одно из наиболее распространенных нейрокожных заболеваний. Как правило, дерматовенеролог становится первым специалистом, который может заподозрить данное заболевание. Несмотря на то что мутация гена при болезни Реклингаузена была выявлена в 17 хромосоме еще в 1987 году, диагностика заболевания до сих пор основывается главным образом на выявлении клинических симптомов. Среди диагностических признаков, рекомендованных Международным комитетом экспертов по нейрофиброматозу, 3 критерия относятся к кожным проявлениям. В дополнение к классическим признакам наличие других элементов, таких как анемический невус и ювенильная ксантогранулема, также могут иметь прогностическое значение. Нейрофиброматоз является мультисистемным заболеванием с разнообразной клинической картиной, меняющейся с возрастом, что обосновывает включение в диспансерное наблюдение не только врача-дерматовенеролога, но и смежных специалистов.
1. Болезни кожи : монография [атлас] / под ред. Н.В. Кунгурова. – Екатеринбург : УрНИИДВиИ, 2014. – 176 с.
2. Еремина М.Г. Пример альтернативного способа коммуникации пациента с нейрофиброматозом в социокультурной среде / М.Г. Еремина, Д.С. Муратова, С.Р. Утц // Саратовский научно-медицинский журнал. – 2014. - Т. 10, № 3. – С. 565-568.
3. Любченко Л.Н. Нейрофиброматоз: генетическая гетерогенность и дифференциальная диагностика / Л.Н. Любченко, М.Г. Филлипова // Саркомы костей, мягких тканей и опухоли кожи. – 2011. - № 4. – С. 29-36.
4. Маратканова Т.В. К вопросу диагностики нейрофиброматоза (клинико-диагностические наблюдения) / Т.В. Маратканова, Г.А. Сташук, Л.Б. Денисова, Л.А. Шерман // Медицинская визуализация. – 2008. - № 6. – С. 114-123.
6. Шнайдер Н.А. Нейрофиброматоз первого типа (болезнь Реклингхаузена) / Н.А. Шнайдер, А.И. Горелов // Сибирское медицинское обозрение. – 2007. - № 3. – С. 91-95.
7. Anderson J.L. Neurofibromatosis type 1 / J.L. Anderson, D.H. Gutmann // Handb Clin Neurol. – 2015. - Vol. 132. – P. 75-86.
8. Barker D. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17 / D. Barker, E. Wright, K. Nguyen, L. Cannon et al. // Science. – 1987. - Vol. 236, № 4805. – P. 1100-1102.
9. Boulanger J.M. Neurofibromatosis type 1 in a pediatric population: Ste-Justine's experience / J.M. Boulanger, A. Larbrisseau // Can J Neurol Sci. – 2005. - Vol. 32, № 2. – P. 225-231.
10. Burkitt Wright E.M. Can the diagnosis of NF1 be excluded clinically? A lack of pigmentary findings in families with spinal neurofibromatosis demonstrates a limitation of clinical diagnosis / E.M. Burkitt Wright, E. Sach, S. Sharif, O. Quarrell et al. // J Med Genet. – 2013. - Vol. 50, № 9. – P. 606-613.
11. Chernoff K.A. Cutaneous and ocular manifestations of neurocutaneous syndromes / K.A. Chernoff, J.V. Schaffer // Clin Dermatol. – 2016. - Vol. 34, № 2. – P. 183-204.
12. Dagalakis U. Puberty and plexiform neurofibroma tumor growth in patients with neurofibromatosis type I / U. Dagalakis, M. Lodish, E. Dombi, N. Sinaii et al. // J Pediatr. – 2014. Vol. 164, № 7. – P. 620-624.
13. Duat Rodriguez A. Phenotypic and genetic features in neurofibromatosis type 1 in children / A. Duat Rodriguez, G.A. Martos Moreno, Y. Martin Santo-Domingo, A. Hernandez Martin et al. // An Pediatr. – 2015. - Vol. 83, № 6. – P. 173-182.
14. Fenot M. Juvenile xanthogranulomas are highly prevalent but transient in young children with neurofibromatosis type 1 / M. Fenot, J.F. Stalder, S. Barbarot // J Am Acad Dermatol. – 2014. Vol. 71, № 2. – P. 389-390.
15. Ferrari F. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1 / F. Ferrari, A. Masurel, L. Olivier-Faivre, P. Vabres // JAMA Dermatol. – 2014. - Vol. 150, № 1. – P. 42-46.
16. Hernández-Martína A. An Update on Neurofibromatosis Type 1: Not Just Café-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease / A. Hernández-Martína, A. Duat-Rodríguezb // Actas Dermosifiliogr. – 2016. - Vol. 107, № 6. – P. 454-464.
17. Hernández-Martín A. An Update on Neurofibromatosis Type 1: Not Just Café-au-Lait Spots and Freckling. Part II. Other Skin Manifestations Characteristic of NF1. NF1 and Cancer / A. Hernández-Martín, A. Duat-Rodríguez // Actas Dermosifiliogr. – 2016. - Vol. 107, № 6. – P. 465-473.
18. Hernandez-Martin A. Nevus anemicus: A distinctive cutaneous finding in neurofibromatosis type 1 / A. Hernandez-Martin, F.J. Garcia-Martinez, A. Duat, I. Lopez-Martin, L. Noguera-Morel // Pediatr Dermatol. – 2015. - Vol. 32, № 5. – P. 342-347.
19. Hirbe A.C. Neurofibromatosis type 1: a multidisciplinary approach to care / A.C. Hirbe, D.H. Gutmann // Lancet Neurol. – 2014. - Vol. 13, № 8. – P. 834-843.
22. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. – 1988. - Vol. 45, № 5. – P. 575-578.
23. Ramos-Geldres T.T. LEOPARD syndrome without hearing loss or pulmonary stenosis: a report of 2 cases / T.T. Ramos-Geldres, P. Dávila-Seijo, A. Duat-Rodríguez, L. Noguera-Morel et al. // Actas Dermosifiliogr. – 2012. - Vol. 106, № 4. – P. 19-22.
24. Raygada M. Juvenile xanthogranuloma in a child with previously unsuspected neurofibromatosis type 1 and juvenile myelomonocytic leukemia / M. Raygada, D.C. Arthur, A.S. Wayne, O.M. Rennert, J.A. Toretsky // Pediatr Blood Cancer. – 2010. - Vol. 54, № 1. – P. 173-175.
25. Rojnueangnit K. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation / K. Rojnueangnit, J. Xie, A. Gomes, A. Sharp et al. // Hum Mutat. – 2015. Vol. 36, № 11. – P. 1052-1063.
26. Ruggieri M. The clinical and diagnostic implications of mosaicism in the neurofibromatoses / M. Ruggieri, S.M. Huson // Neurology. – 2001. - Vol. 56, № 4. – P. 1433-1443.
27. Tadini G. Anemic nevus in neurofibromatosis type 1 / G. Tadini, M. Brena, L. Pezzani // Dermatology. – 2013. - Vol. 115, № 2. – P. 115-118.
28. Takenouchi T. Multiple café au lait spots in familial patients with MAP2K2 mutation / T. Takenouchi, A. Shimizu, C. Torii, R. Kosaki et al. // Am J Med Genet A. – 2014. - Vol. 164A, № 2. – P. 392-396.
29. Zeller J. Blue-red macules and pseudoatrophic macules in neurofibromatosis 1 / J. Zeller, J. Wechsler, J. Revuz, P. Wolkenstein // Ann Dermatol Venereol. – 2002. - Vol. 129, № 2. – P. 180-181.
Нейрофиброматоз представляет собой генетически гетерогенную группу наследственных моногенных заболеваний. В настоящее время описано 7 типов, из которых наибольшее клиническое значение имеют первые два.
Болезнь Реклингаузена (БР, нейрофиброматоз I типа, периферический нейрофиброматоз, НФ 1) относится к группе системных наследственных нейрокутанных факоматозов и характеризуется развитием опухолей эктодермального происхождения. Заболевание было описано в конце XIX века учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. БР встречается с частотой 1:2000 – 1:3500 населения, в России 1,28:10000 [6; 7; 16; 20]. Оба пола болеют одинаково часто. Тип наследования аутосомно-доминантный с высокой пенетрантностью (близкой к 100%) и вариабельной экспрессивностью [5; 6; 20].
Ген НФ 1 был выявлен в 17 хромосоме на локусе q11.2 в 1987 году. Он экспрессируется в шванновских клетках, меланоцитах, лейкоцитах, клетках надпочечников, центральной нервной системы и кодирует белок нейрофибромин [7; 8; 16]. Белок содержит особый домен (НФ1-ГРД), который оказывает тормозное влияние на продукт проонкогена RAS и в норме ингибирует его функцию, за счет чего обеспечивается супрессорный эффект в отношении клеточной пролиферации. Воздействие проонкогена RAS не ограничивается участием в пролиферативных процессах, описано его влияние на формирование когнитивных нарушений в виде затруднения в процессе обучения чтению, письму, математике [6; 8; 18; 20].
Основными диагностическими критериями являются клинические симптомы, рекомендованные Международным комитетом экспертов по нейрофиброматозу, принятые в 1988 году. В соответствии с данными критериями диагноз БР может быть установлен при наличии у больного минимум 2 признаков [3; 4; 22].
1. Наличие 5 и более пигментных пятен цвета «кофе с молоком» диаметром более 5 мм у детей допубертатного возраста и не менее 6 пятен диаметром более 15 мм в постпубертатном возрасте.
2. Веснушчатость в подмышечных и/или паховых складках.
3. Не менее 2 нейрофибром любого типа или одна плексиформная нейрофиброма.
4. Дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных костей с псевдоартрозом или без него.
5. Глиома зрительного нерва.
6. Не менее 2 узелков Лиша (гамартомы) на радужке, выявляемые при исследовании с помощью щелевой лампы.
7. Наличие НФ 1 у родственников первой степени родства.
Из всех диагностических критериев кожные проявления являются наиболее значимыми, так как доступны при осмотре и зачастую бывают первыми симптомами болезни.
Пигментные пятна цвета «кофе с молоком» (франц. «café-at-lait») - первый и постоянный признак болезни Реклингаузена, встречающийся в 95% случаев. Пигментные пятна могут выявляться при рождении, но чаще появляются к трехлетнему возрасту на закрытых участках тела [3; 9; 13]. Они представляют собой монохромные пятна от светло-бежевого до тёмно-коричневого цвета, однородные по структуре, округлых или овальных очертаний, с ровными краями и четкими границами. Размеры могут варьировать от нескольких миллиметров до нескольких сантиметров, однако диагностически значимыми являются пятна диаметром более 5 мм у детей до пубертата и более 15 мм после полового созревания (рис. 1). Размер пятен увеличивается пропорционально росту ребенка [7; 16; 20]. При гистологическом исследовании в очагах обнаруживается повышенное содержание меланина в меланоцитах [2; 9; 13]. Пигментные пятна цвета «кофе с молоком» следует дифференцировать с гиперпигментированными невусами, меланоцитарными невусами, поствоспалительной гиперпигментацией.
Веснушки (симптом Кроува) – это пигментированные пятна размером 1-3 мм, светло-коричневого цвета, локализующиеся в подмышечной и/или паховой области, под молочными железами, как правило, появляющиеся на втором году жизни, реже в первые месяцы после рождения. Солнечное излучение не является провоцирующим фактором их возникновения, в отличие от обычных веснушек. Для их возникновения имеет значение трение и мацерация [2; 3; 5]. Частота выявления веснушек при болезни Реклингаузена варьирует от 21 до 93,7% [9; 13; 16].
Нейрофибромы – это доброкачественные опухоли, производные нервной оболочки периферических нервов. Они состоят из различных типов клеток: шванновских клеток, фибробластов, тучных клеток, эндотелиальных клеток, а также коллагеновых волокон [3; 10; 16]. Нейрофибромы представляют собой округлые узелки на коже и/или в толще кожи, мягкоэластической консистенции, синюшно-красного цвета и/ или цвета нормальной кожи, размерами от просяного зерна до 5 см и более (рис. 1, 2). При пальпации безболезненные, за исключением случаев вовлечения периферических нервов, когда возникают гипостезии и боли, иррадиирующие в соответствующие зоны иннервации [6; 7; 11].
В настоящее время нет единой классификации нейрофибром. M. Ruggieri и соавт. предлагают выделять поверхностные и глубокие образования, а J. Zeller и соавт. подразделяют их на кожные, подкожные и плексиформные [26; 29]. Такое различие происходит вследствие несоответствия клинических проявлений гистологическому расположению нейрофибром. A. Hernández-Martína и соавт. в 2016 году попытались объединить данные классификации. Авторы разделяют нейрофибромы на поверхностные и глубокие, при этом к поверхностным относят кожные (дермальные) и подкожные нейрофибромы, которые расположены в гиподерме, однако также могут иметь дермальный компонент. К глубоким относят плексиформную нейрофиброму [16].
Провоцирующим фактором роста нейрофибром является гормональная перестройка организма: пубертат, беременность. На начальном этапе развития кожные нейрофибромы могут не визуализироваться, в то же время с годами достигать больших размеров, куполообразно возвышаясь над поверхностью кожи. Таким образом, первые видимые элементы появляются в период полового созревания [5; 6; 11].
Нейрофибромы пальпируются как подкожные образования, расположенные линейно по ходу периферических нервов, мягкоэластической консистенции, при пальпации сдвигаются в поперечном направлении вместе с нервным стволом, характерным симптомом является проваливание пальца при легком надавливании (феномен «кнопка звонка»). Могут локализоваться на любом участке тела, однако несколько чаще встречаются на голове и шее, в этом случае их необходимо дифференцировать с увеличенными лимфоузлами [5; 7; 10; 16].
Глубокие плексиформные нейрофибромы обычно присутствуют с рождения и остаются незамеченными при клиническом обследовании до тех пор, пока не появляется неврологическая симптоматика (парестезии, парезы, потеря чувствительности, боль), или же они обнаруживаются при проведении МРТ-исследования [3; 16]. По данным U. Dagalakis и соавт., распространенность плексиформных нейрофибром у детей составляет 10%. У 8–13% больных БР они могут озлокачествляться, перерождаясь в нейрофибросаркому. О злокачественной трансформации свидетельствует непроходящая боль, быстрое увеличение размеров, появление уплотнений, неврологическая симптоматика [3; 12; 19].
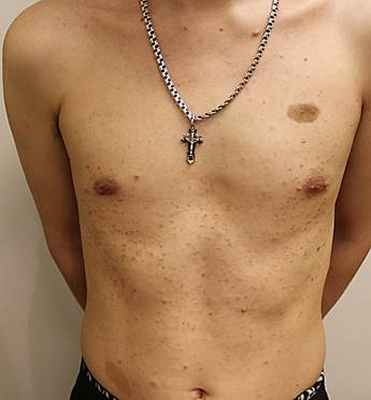
Рис. 1. Пациент А. 18 лет. Болезнь Реклингаузена. Пигментные пятна «кофе с молоком», множественные кожные нейрофибромы на коже передней брюшной стенки
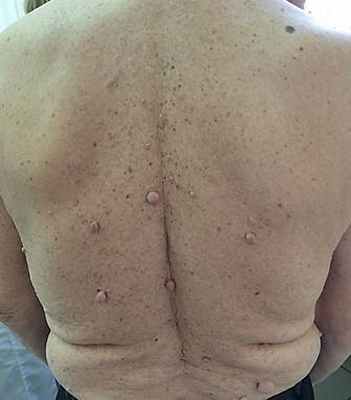
Рис. 2. Пациент В. 47 лет. Множественные кожные нейрофибромы на спине больного нейрофиброматозом I типа
Пятна «кофе с молоком» встречаются не только при БР, но и при других наследственных синдромах и заболеваниях. Дифференциальная диагностика проводится с нейрофиброматозом 2 типа, семейными пятнами café-at-lait, синдромом LEOPARD, синдромом McCune-Albright, синдромом Noonan и другими мультисистемными заболеваниями.
В частности, при нейрофиброматозе 2 типа пигментные пятна цвета «кофе с молоком» встречаются у 80% больных, однако диагностическая ценность их менее значима, чем при БР. Также более характерным является развитие шванном, чем нейрофибром. Из опухолей ЦНС преобладают менингиомы, глиомы, шванномы. Характерна ювенильная задняя субкапсулярная катаракта [5; 6; 16].
Семейный характер пятен café-at-lait должен быть заподозрен при выявлении у родственников разных поколений при отсутствии других проявлений нейрофиброматоза 1 типа. Тип наследования аутосомно-доминантный, однако до сих пор не описаны генетические мутации, приводящие к появлению пигментации [9].
Пигментные пятна у больных с синдромом LEOPARD, как правило, имеют более темную полихромную окраску, полигональные очертания. Кроме кожных проявлений, у пациентов обнаруживаются изменения ЭКГ, глазной гипертелоризм, стеноз легочной артерии, аномалии развития половых органов, нейросенсорная глухота, задержка роста и развития [1; 16; 23].
Для синдрома McCune-Albright характерно наличие пятен «кофе с молоком» бо?льших размеров, чем при БР, с неровными краями, нечеткими границами, полигональных очертаний, часто расположенных с одной стороны, сегментарно. Также у больных выявляется фиброзная дисплазия и гиперфункция эндокринных желез [9; 28].
Пациенты с синдромом Noonan фенотипически похожи на пациентов с нейрофиброматозом 1 типа. Больные, как правило, низкого роста с короткой шеей и треугольным лицом, для них характерно наличие пятен «кофе с молоком», веснушчатость в подмышечных и паховых областях, микроцефалия, птоз, стеноз легочной артерии, снижение интеллекта. Однако при синдроме Noonan не обнаруживаются кожные нейрофибромы, опухоли центральной нервной системы, узелки Лиша [3; 25].
Следует отметить, что характерные клинические признаки БР появляются у больных в разные периоды жизни, и по отдельности не являются патогномоничными, поэтому постановка окончательного диагноза может быть отсрочена на многие годы. А. Hernández-Martína и A. Duat-Rodríguezb в 2016 году сообщают, что в дополнение к классическим проявлениям БР (пятна «кофе с молоком», веснушки и нейрофибромы) у больных часто выявляются другие кожные симптомы, которые могут иметь прогностическую ценность у пациентов с неустановленным диагнозом. К ним относятся: анемический невус, ювенильная ксантогранулема, диффузная гиперпигментация, гипопигментированные пятна [17].
Наиболее часто выявляется анемический невус. Впервые связь между болезнью Реклингаузена и анемическим невусом предположил Naegeli в 1915 г. Анемический невус представляет собой бледное неправильных очертаний пятно размерами от нескольких миллиметров до нескольких сантиметров. Невус может быть одиночным или множественным и располагаться на любом участке, чаще встречается в окологрудинной области. Клинически он не всегда обнаруживается и проявляется после незначительного трения кожных покровов, при этом оставаясь неизменным бледным пятном на фоне общей гиперемии здоровой кожи [11; 17; 27]. M. Marque и соавт. отмечают высокую распространенность анемических невусов у детей, однако зачастую они обнаруживаются в более позднем возрасте при осмотре дерматологом, так как остаются незаметными для родителей. Впоследствии у 50% детей с анемическими невусами был подтвержден диагноз «болезнь Реклингаузена» [17; 21].
Ювенильная ксантогранулема является наиболее распространенной формой гистиоцитоза. Она представляет собой папулу округлой или овальной формы, плотноватой консистенции, размерами от 1 до 2 см, желтоватого или красно-желтого цвета, безболезненная при пальпации. Характерна спонтанная инволюция в пубертатном периоде. Ювенильные ксантогранулемы могут быть единичными и множественными. Локализуются чаще всего на лице, волосистой части головы, туловище, реже на слизистых оболочках. По данным M. Fenot (2014), распространенность ювенильной ксантогранулемы у больных БР варьирует от 0,7% у взрослых до 37,5% у детей, особенно у детей в возрасте до 9 лет [14; 15; 24].
Также у пациентов с болезнью Реклингаузена может встречаться зуд, диффузная гипер- и/ или гипопигментация. Зуд беспокоит 20% больных БР и существенно снижает их качество жизни. Диффузная гиперпигментация отчетливо выявляется при сравнении тона кожи больного с тоном кожи других здоровых членов семьи [7; 17].
Нейрофиброматоз опасен прежде всего своими осложнениями, возникающими при несвоевременной диагностике, что обуславливает необходимость диспансерного наблюдения. Наиболее прогностически неблагоприятным осложнением БР является озлокачествление имеющихся нейрофибром. Также к осложнениям НФ 1 относятся глиома зрительных нервов, приводящая к слепоте, феохромоцитома с развитием симптоматической злокачественной артериальной гипертензии. При БР описаны васкулиты, приводящие к стенозу почечных артерий, коарктации аорты [6; 10; 11].
Таким образом, ранняя диагностика и диспансеризация больных нейрофиброматозом имеет решающее значение в прогнозе и качестве жизни пациентов. Повышение знаний об указанной патологии у специалистов разного профиля будет способствовать своевременной диагностике НФ 1 и выявлению осложнений. Болезнь Реклингаузена является одним из заболеваний, где важна преемственность в работе врачей разных специальностей.
Читайте также:
- Резистивный синдром. Проявления резистивного синдрома
- Симпатический ствол. Топография симпатического ствола. Ветви симпатического ствола.
- Неконкурентное необратимое ингибирование. Аллостерические ферменты.
- Симптомы ангиофибромы носоглотки и ее лечение
- Сфигмография. Флебография. Анакрота. Катакрота. Флебограмма.