
Лейкоэнцефалопатия с субкортикальными кистами
1. Сокращения:
• Мегалэнцефалия с лейкоэнцефалопатией и субкортикальными кистами (МЛК)
2. Синонимы:
• Ранее используемая терминология
о Вакуолирующая мегалоэнцефалическая лейкоэнцефалопатия с доброкачественным, медленно прогрессирующим течением
о Лейкоэнцефалопатия с дебютом в младенчестве, отеком и непропорционально легким течением
о Болезнь Ван-дер-Кнаапа:
- Одно из многих заболеваний с именными названиями
о Мегалоэнцефалическая лейкодистрофия у индийцев сообщества Агравал
3. Определение:
• Аутосомно-рецессивное нарушение работы регулируемых объемом анионных каналов в астроцитах:
о Характеризуется макроцефалией, ухудшением двигательного/когнитивного статуса, атаксией, мышечной спастичностью
1. Общие характеристики мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Лучший диагностический критерий:
о Отек, изменения диффузного характера со стороны белого вещества (БВ) больших полушарий
о Субкортикальные кисты преимущественно локализуются в передних отделах височных, а также в лобно-теменных областях
• Локализация:
о Диффузное вовлечение в процесс БВ, включая субкортикальные 11-волокна:
- Субкортикальные кисты:
Наиболее часто: передние отделы височных областей
Также часто: лобно-теменные области
- ± вовлечение задних отделов внутренних капсул
- Минимальное вовлечение в процесс БВ мозжечка
• Размеры:
о С течением времени кисты увеличиваются в размерах и количестве
2. КТ признаки мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Бесконтрастная КТ:
о ↓ плотности вовлеченного в процесс БВ
• КТ с контрастированием:
о Отсутствие накопления контраста
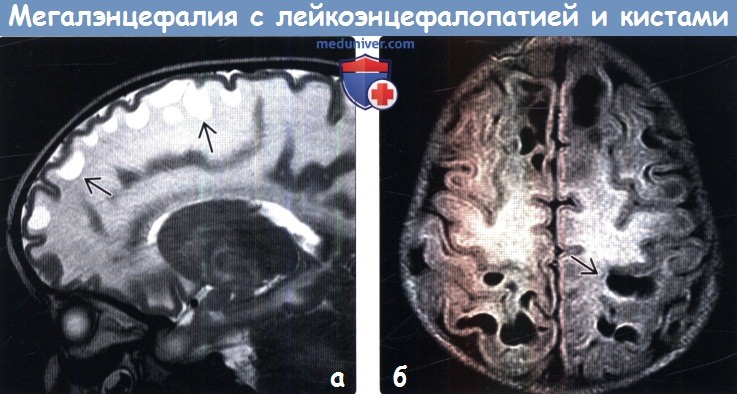
(а) МРТ, Т2-ВИ, сагиттальный срез: у двухлетнего ребенка с мегалэнцефалией и замедлением процесса достижения контрольных показателей развития определяется отек белого вещества, а также протяженная кистозная трансформация субкортикального белого вещества лобной доли.
(б) MPT, FLAIR, аксиальный срез: у этого же двухлетнего пациента определяются аномальное повышение интенсивности сигнала от белого вещества вследствие гипомиелинизации, а также множественные крупные субкортикальные кисты в лобных и теменных областях.
3. МРТ признаки мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Т1-ВИ:
о ↓ интенсивности сигнала от вовлеченного в процесс БВ
• Т2-ВИ:
о ↑ интенсивности сигнала от вовлеченного в процесс БВ
- Белое вещество больших полушарий:
Относительная сохранность мозолистого тела
- ± вовлечение в процесс дорсальной 1/3 заднего бедра внутренней капсулы
- ± вовлечение в процесстрактов БВ ствола мозга в младшем возрасте
• FLAIR:
о ↑ интенсивности сигнала от вовлеченного в процесс БВ
о Субкортикальные кисты:
- Наиболее часто в передних отделах височных областей и лобных областях
- Сигнал от кист схож с сигналом от СМЖ
• ДВИ:
о DTI: ↓ анизотропии, ↑ значений ИКД
- Обусловлено ↓ количества воды в интерстициальном пространстве
• Постконтрастные Т1-ВИ:
о Отсутствие накопления контраста
о Вероятно, в контрастном усилении нет необходимости
• МР-спектроскопия:
о ↓ всех метаболитов в области кистозной трансформации
о ↓ пика NAA в БВ
о Нормальный пик миоинозитола
о ± сигнал от лактата
4. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ с МР-спектроскопией:
- ± контрастное усиление (для исключения лейкодистрофий, характеризуемых накоплением контраста)
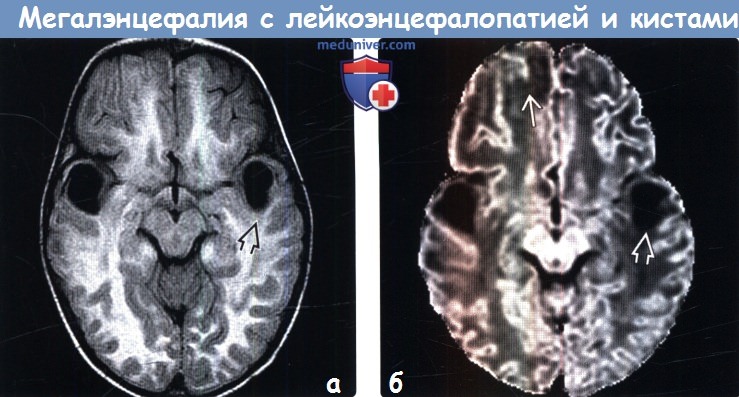
(а) MPT, FLAIR, аксиальный срез: у ребенка 22 месяцев с макроцефалией и замедлением процесса достижения контрольных показателей развития на фоне отечного и аномально гиперинтенсивного белого вещества в структуре височных долей визуализируются крупные кисты, что типично для мегалинцефалии с лейкоэнцефалопатией и кистами (МЛК).
(б) МРТ, ДВИ, аксиальный срез: у этого же 22-месячного ребенка подтверждается выраженное повышение диффузии в кистах и, в меньшей степени, в субкортикальном белом веществе.
в) Дифференциальная диагностика мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
2. Другие лейкодистрофии, характеризующиеся накоплением контраста:
• Болезнь Александера:
о Аномальный сигнал + контрастное усиление БВ лобных долей, а также эпендимальных поверхностей
о Вовлечение в процесс базальных ядер
• Х-сцепленная адренолейкодистрофия:
о Аномальный сигнал и контрастное усиление перитригонального БВ и валика мозолистого тела
1. Общие характеристики мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Этиология:
о Врожденные генетические ошибки
о Нарушение водного гомеостаза и осмотического баланса вследствие нарушения функции регулируемых по объему анионных каналов (VRAC):
- VRAC в астроцитах реагируют на изменения осмолярности внеклеточной жидкости
- Отражает значимые изменения объема клеток как компонента процесса осморегуляции
- MLC1 играет важную роль в активности VRAC; GlialCAM-это шаперон MLC1 в ионных каналах
• Генетика:
о Аутосомно-рецессивное наследование; локус гена: 22q(tel)
- Множество различных мутаций генов MLC1 и GLIALCAM:
- Мутации, распределенные по целым генам, типам включают:
Мутации сайта сплайсинга
Нонсенс-мутации
Миссенс-мутации
Делеции и инсерции
- Мутации идентифицируются в 80% случаев; подозрителен второй локус
- Большинство мутаций имеют частный характер
- Эффект основателя встречается в популяционных субизолятах
• Ассоциированные аномалии:
о MLC1 в ЦНС экспрессируется в концевых отростках астроцитов на уровне гемато-энцефалического и ликворо-энцефалического барьеров:
- GlialCAM является шапероном для MLC1 в ионных каналах астроцитов
о MLC1 также экспрессируется в лейкоцитах периферической крови, селезенки:
- Однако системный процесс или вовлечение других органов отсутствуют
2. Макроскопические и хирургические особенности:
• Спонгиоформная лейкоэнцефалопатия:
о Вакуолизация субкортикального белого вещества
3. Микроскопия:
• Расщепление миелина на межпромежуточной линии
• Вакуолизация самых наружных слоев миелиновых оболочек
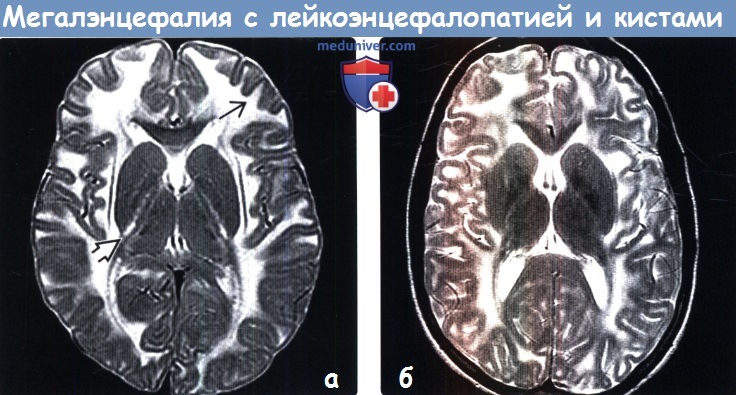
(а) МРТ, Т2-ВИ, аксиальный срез: у шестилетнего ребенка с мегалэнцефалией с лейкоэнцефалопатией и кистами (МЛК) отмечается менее выраженный отек белого вещества. Тем не менее отмечается сохранение аномального повышения интенсивности сигнала со стороны задних бедер внутренних капсул и субкортикальных U-волокон.
(б) МРТ, Т2-ВИ, аксиальный срез: у пациента 14 лет с мегалэнцефалией с лейкоэнцефалопатией и кистами (МЛК) определяется расширение борозд вследствие атрофии, что является частой находкой при прогрессировании заболевания. Обратите внимание на сохранение повышения интенсивности сигнала от белого вещества большого мозга с вовлечением субкортикальных U-волокон.
г) Клиническая картина:
1. Проявления мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Наиболее частые признаки/симптомы:
о Макроцефалия при рождении или в течение первого года жизни
о Отсроченное развитие медленно прогрессирующих двигательных нарушений (ухудшение когнитивного статуса характеризуется еще более медленным прогрессированием) несмотря на выраженные изменения со стороны МРТ:
- Развитие в течение первого года жизни часто в норме
• Другие признаки/симптомы:
о Редко: ранняя манифестация в виде задержки развития
о Редко кратковременная кома после легкой травмы головы
• Клинический профиль:
о Макроцефалия
о Очень медленное ухудшение когнитивного статуса:
- В 50% случаев возможны проблемы с обучением
о Мозжечковая атаксия и вовлечение пирамидных трактов
о Ухудшение двигательного статуса:
- Утрата способности ходить на поздних стадиях
- Иногда задержка автономной ходьбы
о Легкая травма головы → судороги и временное ухудшение состояния
2. Демография:
• Возраст:
о Макроцефалия в возрасте до первого года жизни
• Этническая принадлежность:
о Увеличение численности населения изолятов
- Часто мутации гена MLC встречаются в:
Отдельном индийском сообществе (Агарвал)
Сообществе ливийских евреев
Турецкие сообщества
Некоторые японские семьи из-за эффекта основателя
- Мутация сообщества Агарвал:
Инсерция (с.135_136insC) — часто, встречается фенотипическая изменчивость
• Эпидемиология:
о Встречается редко
о Частота носительства в некоторых популяциях с высоким уровнем родства составляет до 1/40
3. Течение и прогноз:
• Раннее развитие отека белого вещества:
о Степень выраженности раннего отека белого вещества со временем уменьшается
о В результате развивается атрофия
• Клинические проявления прогрессируют медленно
4. Лечение мегалэнцефалии с лейкоэнцефалопатией и кистами (МЛК):
• Купирование симптомов (судороги, мышечная спастичность)
• Пренатальная диагностика возможна в семьях с известными мутациями
е) Список литературы:
- Kariminejad A et al: Eight novel mutations in MLC1 from 18 Iranian patients with megalencephalic leukoencephalopathy with subcortical cysts. Eur J Med Genet. 58(2)71-4, 2015
- Mahmoud IG et al: Clinical, neuroimaging, and genetic characteristics of megalencephalic leukoencephalopathy with subcortical cysts in Egyptian patients. Pediatr Neurol. 50(2):140-8, 2014
- Capdevila-Nortes X et al: Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum Mol Genet. 22(21):4405-16, 2013
- Rodriguez D: Leukodystrophies with astrocytic dysfunction. Handb Clin Neurol. 113:1619-28, 2013
- Renaud DL: Leukoencephalopathies associated with macrocephaly. Semin Neurol. 32(1 ):34-41, 2012
- van der Knaap MS et al: Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 11(11):973—85, 2012
- Miles L et al: Megalencephalic leukoencephalopathy with subcortical cysts: a third confirmed case with literature review. Pediatr Dev Pathol. 12(3)480-6, 2009
- Boor I et al: MLC1 is associated with the dystrophin-glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 114(4):403—10, 2007
- Kiriyama T et al: SPECT revealed cortical dysfunction in a patient who had genetically definite megalencephalic leukoencephalopathy with subcortical cysts. Clin Neurol Neurosurg. 109(6):526—30, 2007
- Teijido О et al: Expression patterns of MLC1 protein in the central and peripheral nervous systems. Neurobiol Dis. 26(3):532-45, 2007
- Ilja Boor PK et al: Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum Mutat. 27(6):505—12, 2006
- Morita H et al: MR imaging and 1H-MR spectroscopy of a case of van der Knaap disease. Brain Dev. 28(7):466-9, 2006
Редактор: Искандер Милевски. Дата публикации: 25.4.2019
Лейкоэнцефалопатия – это такое заболевание, которое приводит к повреждению белого вещества, составляющего основу головного мозга, что провоцирует многочисленные неврологические синдромы. Характеризуется быстрым прогрессированием и развитием демиелинизирующих патологических процессов.
Определение патологии
Лейкоэнцефалопатия всегда сопровождается поражением белого мозгового вещества, которое возникает вследствие разных причин: сосудистых патологий, инфицирования полиомавирусом, генных мутаций. Инфекционная форма развивается из-за реактивации полиомавируса JC, который в латентном состоянии находится в организме многих людей.
Наследственная форма заболевания аутосомно-рецессивного генеза возникает на фоне генной мутации (преимущественно гена EIF2B5). Мелкоочаговая лейкоэнцефалопатия сосудистого генеза – это такое заболевание, которое развивается вследствие поражения небольших элементов кровеносной системы мозга, что провоцирует характерные для гипоксически-ишемического поражения тканей симптомы.
Классификация заболевания
Различают формы патологии в зависимости от причин возникновения. Сосудистая форма лейкоэнцефалопатии, известная как болезнь Бинсвангера – это такое заболевание головного мозга, которое носит прогрессирующий характер, развивается на фоне гипертонической болезни, что указывает на ведущую роль в патогенезе атеросклероза и других патологий сосудов.
Клиническая картина напоминает проявления субкортикальной (подкорковой) энцефалопатии артериосклеротической этиологии. Патогенез связан с поражением белого вещества на фоне атеросклероза мелких артерий и артериол, вследствие чего происходит утолщение сосудистых стенок и сужение просвета, развивается гиалиноз – белковая дистрофия с замещением нормальной ткани более плотной, малоэластичной структурой.
Типичные осложнения: ТИА (преходящие нарушения мозгового кровообращения), инсульты. Третья часть всех клинических случаев сосудистой деменции обусловлена мультифокальной лейкоэнцефалопатией. В 80% случаев возраст пациентов составляет 50-70 лет. Очаговое поражение белого вещества инфекционного генеза возникает вследствие реактивации вируса JC.
Перивентрикулярная лейкоэнцефалопатия, известная как перивентрикулярная лейкомаляция, характеризуется возникновением очагов поражения белого вещества чаще возле желудочковой системы. При масштабном поражении очаги распространяются в центральные отделы белого вещества.
Демиелинизирующий процесс агрессивного, длительного течения приводит к образованию некротических полостей в мозговых структурах. Лейкоэнцефалопатия (лейкомаляция) головного мозга, обнаруженная у детей, часто является причиной развития в детском возрасте церебрального паралича (ДЦП).
Причины возникновения
Основная причина развития формы инфекционного генеза – реактивация полиомавируса. Это безоболочечный вирус JC, который обнаруживается у 80% населения. Обычно не вызывает развитие заболеваний. В этом случае инфицированные люди являются носителями. Активация происходит на фоне подавления деятельности иммунной системы, часто у пациентов с диагнозом ВИЧ.
Инфицирование ВИЧ часто сопровождается типичным симптомом – развитием многоочаговой лейкоэнцефалопатии с прогрессирующим течением патологии. Заболевание возникает как результат терапии иммуномодуляторами (после имплантации органов), иммуносупрессорами или моноклональными антителами, из-за чего происходит угнетение иммунной системы.
Терапия при помощи моноклональных антител проводится в отношении рассеянного склероза, аутоиммунных заболеваний крови, неходжкинской лимфомы, артрита ревматоидной формы. Вирус JC попадает в организм через органы желудочно-кишечного тракта с зараженной водой и пищей или воздушно-капельным путем. Процесс инфицирования протекает бессимптомно.
Вирус пребывает в организме латентно до появления факторов, провоцирующих активацию. Провоцирующие факторы ассоциируются с пересадкой стволовых клеток и лечением препаратами – аналогами пурина (Фопурин, Меркаптопурин, Пуринетол). Мелкоочаговая лейкоэнцефалопатия, которая развилась на фоне сосудистых патологий, обычно спровоцирована факторами:
- Артериальная гипертензия, длительно протекающая, устойчивая.
- Артериальная гипотензия.
- Патологическое изменение циркадного (биологического) ритма артериального давления – резкое повышение или понижение показателей давления в ночное время.
Патологические изменения в мозговых структурах происходят в результате хронической гипоксии – кислородного голодания. Вероятные причины включают врожденные аномалии развития элементов сосудистой системы, возрастные деформации, посттравматические дефекты и другие нарушения, провоцирующие ухудшение церебрального кровотока.
Симптоматика
Мелкоочаговая энцефалопатия инфекционного генеза характеризуется отсутствием признаков воспалительного процесса в мозговом веществе. Проникая в ЦНС вирус вызывает лизис (растворение) олигодендроцитов, что приводит к масштабной демиелинизации – повреждению миелиновых оболочек.
Инфицированные олигодендроциты располагаются по краям очага демиелинизации. Постепенно патологический процесс охватывает обширные участки мозга, что провоцирует нарастающее развитие неврологической симптоматики:
- Гемипарезы (парезы в одной стороне тела).
- Гемисоматосенсорные расстройства (нарушение чувствительности – онемение, покалывание, изменение восприятия температуры, изменение реакции на болевые раздражители, искажение пространственного представления о положении отдельных частей тела относительно друг друга).
- Эпилептические припадки. Наблюдаются у 20% больных, что указывает на близость очага к коре.
В зависимости от локализации очага поражения появляются такие симптомы, как атаксия (нарушение согласованности при сокращении группы мышц), афазия (речевая дисфункция), зрительные расстройства, апраксия (нарушение целенаправленных движений), дисметрия (избыточность или недостаточность при выполнении произвольных движений). Для патологии типичны когнитивные расстройства, нередко перерастающие в деменцию.
Поражение тканей полушарий с соответствующей симптоматикой в клинической практике встречается в 10 раз чаще, чем стволовых структур. В ходе исследования МРТ обнаруживаются крупные субкортикальные (подкорковые) очаги – гиперинтенсивные (режим Т2) и гипоинтенсивные (режим Т1). С увеличением диаметра очага неврологический дефицит нарастает. Сосудистая лейкоэнцефалопатия сопровождается симптомами:
- Когнитивные расстройства (ухудшение памяти и умственной деятлеьности) с тенденцией к прогрессированию.
- Атаксия, моторная дисфункция.
- Нарушение двигательной координации.
- Недержание мочи, непроизвольная дефекация.
Для поздних стадий течения заболевания свойственно развитие слабоумия. Больные теряют способность к самообслуживанию, круглосуточно нуждаются в медицинской помощи. У больных отсутствует интерес к речевой, двигательной, познавательной, психической деятельности. Периодически возникает чувство эйфории. У некоторых пациентов происходят эпилептические припадки.
Для болезни Бинсвангера типична тенденция к прогрессированию. Нередко наблюдаются длительные периоды стабильного состояния. Слабоумие развивается из-за нарушения корково-подкорковых связей, которые возникают вследствие повреждения белого вещества. Не последнюю роль в патогенезе играет дисфункция таламуса и базальных ганглиев.
Очаговая лейкоэнцефалопатия – это такое заболевание, для которого характерно изменение походки, что указывает на поражение сосудистого генеза экстрапирамидной системы. Походка больного становится медленной, семенящей, шаги укорачиваются, наблюдается постуральная дисфункция – затруднение движения на поворотах. Пациенту сложно инициировать (начать) движение. Иногда двигательные расстройства проявляются по типу паркинсонизма (ригидность, скованность мышц, тремор).
Симптоматика дополняется гемипарезами (парез в одной половине тела) и псевдобульбарным синдромом (дизартрия – нарушение произношения, дисфония – ослабление силы голоса, дисфагия – затруднения при глотании, непроизвольный плач или смех). Для постановки диагноза многоочаговая лейкоэнцефалопатия сосудистой этиологии необходимо наличие признаков:
- Деменция.
- Факторы риска развития сосудистых заболеваний или признаки сосудистой патологии, затронувшей кровеносную систему мозга.
- Неврологические синдромы, характерные для поражения субкортикальных (подкорковых) структур мозга (изменение походки, недержание мочи, паратония – непроизвольное сопротивление пассивным движениям).
Исследование КТ показывает двухсторонний лейкоареоз – малую ишемию сосудов, повреждения элементов сосудистой системы белого вещества. При перивентрикулярной лейкоэнцефалопатии на МР-томограмме различается диффузно-очаговое поражение белого вещества преимущественно симметричное.
Для сравнения при вирусном поражении очаги расположены преимущественно ассиметрично. Единичный неспецифический очаг лейкоэнцефалопатии может быть ошибочно расценен, как проявление инсульта.
Методы диагностики
Ранняя диагностика JС-ассоциированной инфекции позволяет своевременно начать лечение. Нейровизуализация остается приоритетным способом обнаружения участков патологически измененной ткани. Основные методы инструментальной диагностики:
- МРТ, КТ.
- Допплерография сосудов мозга.
- Электроэнцефалография.
- Биопсия. Исследование мозгового биоптата. Чувствительность метода оценивается в 64-90%.
Исследование типа ПЦР помогает обнаружить ДНК вируса. Чувствительность метода оценивается в 72-92%.
Способы лечения
Лечение патологии сосудистого генеза предполагает устранение причин, спровоцировавших развитие заболевания. Чаще назначают лекарства и другие методы терапии, направленные на стабилизацию повышенного или пониженного артериального давления. Показаны препараты:
- Улучшающие микроциркуляцию крови в мозговых структурах.
- Стимулирующие метаболические процессы в клетках мозга.
- Устраняющие неврологическую симптоматику.
Параллельно проводится терапия атеросклероза и сопутствующих соматических болезней. Специфического лечения при вирусной форме не существует. Эффективными мерами считаются:
- Прекращение приема иммуносупрессоров (кортикостероиды, цитостатики). Или уменьшение их дозировки.
- Отмена Натализумаба – препарата на основе моноклональных антител, если реактивация вируса произошла вследствие его применения. Параллельно рекомендуется проведение плазмафереза до 5 сеансов ежедневно для выведения препарата.
- Назначение Мефлоцина (антималярийный препарат). Некоторые исследования показывают эффективность лекарства, которое замедляет репликацию (размножение) вируса JС.
Антидепрессант Миртазапин ингибирует обратный захват серотонина, препятствует распространению вируса JС. Механизм действия основан на блокировании рецепторов 5-НТ2, которые являются мишенью для полиомавируса.
Прогноз
Сколько живут при диагнозе лейкоэнцефалопатия головного мозга, зависит от причин развития и характера течения патологии. Прогноз продолжительности жизни составляется индивидуально лечащим врачом с учетом возраста и физического состояния пациента. Прогноз заболевания сосудистого генеза условно благоприятный. Своевременная коррекция сосудистых нарушений позволяет отсрочить масштабное повреждение белого вещества.

При диагнозе ВИЧ лейкоэнцефалопатия, обусловленная вирусом, занимает второе место среди причин смертности, уступая только неходжкинской лимфоме. Многочисленные результаты аутопсии показали отсутствие прямой связи между смертностью и морфологическими особенностями строения очагов (размеры, локализация, степень атрофии тканей, гидроцефалия) при вирусной форме болезни.
Профилактические мероприятия
Для профилактики патологии больным рассеянным склерозом, которым проводится терапия Натализумабом, назначают превентивное обследование на наличие вируса JС. Профилактические меры включают общие рекомендации по укреплению иммунитета:
- Закаливающие процедуры.
- Отказ от вредных привычек.
- Организация здорового, полноценного питания.
- Активный образ жизни, дозированные физические нагрузки.
Предотвращение родовых травм в перинатальный период и повреждений в области головы во взрослом возрасте, систематический контроль и коррекция значений артериального давления помогут избежать осложнений и быстрого прогрессирования заболевания.
Лейкоэнцефалопатия – прогрессирующий процесс разрушения белого вещества. Ранняя диагностика и корректная терапия патологии сосудистого генеза способствует значительному улучшению состояния больного.
У 5% пациентов с энцефалопатией Ван дер Кнаап не удаётся обнаружить ни одного из вышеперечисленных генетических дефектов.
Уже при рождении (а чаще, на первом году жизни) становится заметной макроцефалия, которая присутствует практически у всех пациентов. После года рост окружности головы замедляется и вскоре размеры её начинают приближаться к норме. Вначале умственное и физическое развитие как правило соответствуют возрастной норме, однако, постепенно становится заметным несущественное отставание (умственное развивается позже и протекает мягче физического).

Внешний вид ребёнка с энцефалопатией Ван дер Кнаап (источник: Abdel-Salam G.M.H., Abdel-Hamid M.S., Ismail S.I. et al. Megalencephalic leukoencephalopathy with cysts in twelve Egyptian patients: novel mutations in MLC1 and HEPACAM and a founder effect // Metab. Brain Dis., 2016. – Vol.31. – P.1171-1179)
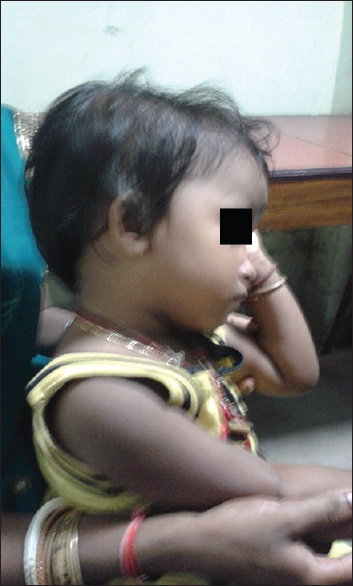
Внешний вид ребёнка с энцефалопатией Ван дер Кнаап (источник: Bhattacharyya K.B., Rai S. Megaencephalic leukoencephalopathy with subcortical cysts in a young Bengali girl // Neurol. India, 2015. – Vol.63. – P.436-743)
Когда ребёнок начинает ходить, становятся заметными нарушения походки – она неуверенная, дети часто падают. Мышечный тонус при этом имеет тенденцию к понижению, за исключением тонуса стоп, который обычно повышен. В первые годы жизни отмечается медленное прогрессирование нарушений моторики, однако, на первый план постепенно начинает выходить статическая и динамическая атаксия, атаксия походки. Также постепенно присоединяется дизартрия, иногда – с дисфагией. Появляются пирамидные знаки, спастичность, реже – экстрапирамидные нарушения (дистония, атетоз, тики). Способность к самостоятельному передвижению постепенно утрачивается, и к 10-20 годам многие пациенты становятся прикованы к инвалидной коляске; в более благоприятных случаях она сохраняется до 50 лет.
Иногда возникают поведенческие расстройства, аутизм. Очень характерны эпилептические приступы, развивающиеся на первом году жизни; обычно они хорошо поддаются лечению, но нередки (15-20%) эпилептические статусы, как правило, развивающиеся в течение нескольких лет после дебюта эпилептического синдрома. Даже малейшая травма головы может приводить к временному нарастанию клинических проявлений, в особо тяжёлых случаях – вплоть эпилептического статуса или до коматозного состояния.
Выделяют 2 разновидности фенотипической картины заболевания – классическую (варианты MLC1 и MLC2A) и благоприятную (вариант MLC2B). По сравнению с вышеописанными проявлениями классического фенотипа, благоприятный вариант характеризуется более мягкой манифестацией и улучшением двигательных функций после года жизни, более редким развитием эпилептического синдрома (лишь у 10% пациентов).
У родителей пробанда – носителей мутаттного гена – может наблюдаться макроцефалия без нарушений двигательных и когнитивных функций, в некоторых случаях – когнитивные/поведенческие расстройства и неловкость движений.
Типичными МРТ-признаками классической формы являются: 1) мегалэнцефалия; 2) диффузное двухстороннее симметричное поражение полушарного белого вещества (гиперинтенсивное в режиме Т2 и гипоинтенсивное в режимеТ1) с признаками его незначительного отёка; 3) относительная сохранность центральных структур (мозолистого тела, внутренней капсулы, ствола мозга); 4) относительно несущественное поражение белого вещества мозжечка без признаков отёка; 5) практически патогномоничное наличие субкортикальных кист в передних отделах височных долей, а также зачастую – и лобно-теменных областях; 6) регресс отёчности белого вещества со временем, приводящий к появлению картины атрофии мозга; 7) повышенная проницаемость отёчного белого вещества в режиме DWI (диффузионно-взвешенных изображений).
В некоторых случаях субкортикальные кисты могут увеличиваться в размерах и количестве. Иногда они достигают больших размеров, распространяясь на значительные объёмы белого вещества лобно-теменных областей. Возможен также и регресс патологических изменений белого вещества.
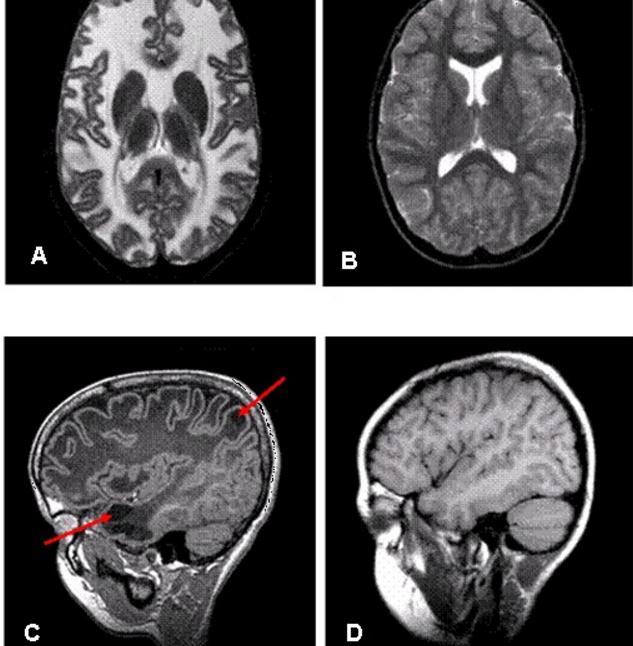
МРТ-картина энцефалопатии Ван дер Кнаап (рис. А,С) и нормального головного мозга (рис.B, D). А – Т2-взвешенное поперечное изображение головного мозга 9-летнего ребёнка с энцефалопатией Ван дер Кнаап: видно диффузное поражение белого вещества полушарий с признаками его отёка; С – Т2-взвешенное сагиттальное изображение головного мозга того же ребёнка, на котором видны субкортикальные кисты в передневисочной и теменной областях (показаны стрелками) (источник: Van der Knaap MS., Abbink T.E.M., Min R. Megalencephalic Leukoencephalopathy with Subcortical Cysts. Synonym: Van der Knaap Disease. – in: Adam M.P., Ardinger H.H., Pagon R.A. et al., eds. Gene Reviews // Seattle (WA): University of Washington, Seattle; 1993-2018)
МРТ-картина благоприятной формы энцефалопатии Ван дер Кнаап на первом году не отличается от таковой при классической форме (за исключением разве что практически интактного белого вещества мозжечка); в последующем наблюдается существенный регресс патологических изменений – через несколько лет головной мозг может выглядеть практически нормальным, с минимальным поражением белого вещества лобной и теменной областей и сохранившимися субкортикальными кистами в передневисочных областях.
Верификацию диагноза можно осуществить методом секвенирования – вначале производится секвенирование гена MLC1, затем, при отрицательном результате – гена HEPACAM (при благоприятном варианте вначале проводится анализ гена HEPACAM). Если патогенетический вариант не найден, возможно применение метода поиска делеции/дупликации в этих генах.
Описаны случаи повышения уровня глицина в ликворе
Дифференциальный диагноз проводится с рядом заболеваний, ядром клинической картины которых является макроцефалия с диффузной лейкоэнцефалопатией: болезнью Канаван(–Ван-Богарта – Бертрана), болезнью Александера, GM2-ганглиозидозами (I типа – болезнь Тея – Сакса, II типа – болезнь Зандхоффа(–Яцкевича(–Пильца)), GM1-ганглиозидозами (ранний и поздний инфантильные типы), L-2-оксиглутаровой ацидурией. Кроме того, некоторые случаи мерозин-дефицитной врождённой мышечной дистрофии типа 1А могут протекать с макроцефалией. Детали клинической картины и течение этих заболеваний, как правило, всё же имеют отличия; МРТ-картина ни одного из них не включает все признаки энцефалопатии Ван дер Кнаап. Если к году жизни окружность головы больного ребёнка существенно превышает норму, то с весьма высокой вероятностью у него нет энцефалопатии Ван дер Кнаап.
Прогноз для жизни при этом заболевании в целом благоприятный, хотя описаны случаи смерти в возрасте 10-20 лет. Если пациент сохраняет способность к ходьбе, самостоятельной или с поддержкой, к 15 годам, то скорее всего он будет оставаться ходячим и в последующем.
Патогенетического лечения до настоящего времени не разработано. Применение таких препаратов, как диуретики, ацетазоламид, креатинина моногидрат не продемонстрировало эффективности. Показана физиотерапия, логопедическая помощь, обучение по индивидуальному плану, антиконвульсанты при развитии эписиндрома. Учитывая высокий риск осложнений при травмах головы в потенциально травматичных ситуациях рекомендуется ношение шлема.
Читайте также: