
Фолликулотропный вариант грибовидного микоза
Первичные Т-клеточные кожные лимфомы представляют собой гетерогенную группу лимфопролиферативных заболеваний, характеризующуюся клональной пролиферацией Т-лимфоцитов в коже. Они составляют 75–80 % всех кожных лимфом. Разграничение первичных лимфом кожи и
Первичные Т-клеточные кожные лимфомы представляют собой гетерогенную группу лимфопролиферативных заболеваний, характеризующуюся клональной пролиферацией Т-лимфоцитов в коже. Они составляют 75–80 % всех кожных лимфом. Разграничение первичных лимфом кожи и вторичных ее поражений при других лимфомах требует тщательного анализа клинической картины, патологических, иммунологических и молекулярно-генетических данных. Первичные лимфомы кожи отличаются от нодальных лимфом характером течения, прогнозом и подходами терапии. Это обстоятельство вызвало необходимость создания подробной классификации, отражающей весь спектр первичных кожных лимфом. В 2005 г. на основе классификации ВОЗ для опухолей гемопоэтической и лимфоидной тканей [1] и классификации лимфом кожи Европейской организации по исследованию и лечению рака (EORTC) [2] создана ВОЗ/EORTC-классификация кожных лимфом, наиболее полно охватывающая весь спектр этих заболеваний [3].
Т- и NK-клеточные лимфомы кожи
-
Грибовидный микоз (ГМ):
— синдром гранулематозной вялой кожи.
— первичная анапластическая крупноклеточная лимфома кожи;
Самыми распространенными подтипами являются: ГМ, синдром Сезари, первичная анапластическая крупноклеточная лимфома кожи и лимфоматоидный папулез. Они составляют приблизительно 95 % всех Т-клеточных кожных лимфом.
Гистологическая картина на ранних стадиях ГМ неспецифична и может быть схожа с таковой при доброкачественном воспалительном дерматозе: отмечаются периваскулярные инфильтраты в сочетании с псориазоформной гиперплазией эпидермиса [5]. Для бляшечной стадии характерен плотный полосовидный инфильтрат в верхней части дермы, содержащий высокий процент церебриформных лимфоцитов с выраженным эпидермотропизмом. Внутриэпидермальные скопления атипичных лимфоцитов (микроабсцессы Потрие) являются характерной чертой этой стадии, но встречаются лишь в 10 % случаев [6]. С прогрессированием в опухолевую стадию эпидермотропизм исчезает, а инфильтрат, состоящий из церебриформных лимфоцитов малых, средних и крупных размеров, становится диффузным и может проникать в подкожную жировую клетчатку.
Опухолевые клетки при ГМ имеют фенотип зрелых Т-лимфоцитов памяти (CD3 + , CD4 + , CD45RO + , CD8 - ). Редко может наблюдаться фенотип CD4 - , CD8 + . Клиническое течение и прогноз в таких случаях не отличаются от классического варианта, и, следовательно, их не следует рассматривать отдельно. О наличии аберрантного фенотипа при ГМ говорит утрата пан-Т-клеточных антигенов CD2, CD3, CD5, CD7, что во многих случаях является важным дополнением к диагнозу [7].
Прогноз при ГМ напрямую зависит от стадии заболевания, характера и распространенности кожного процесса, а также наличия внекожных поражений.
В 1978 г. Национальным институтом рака США была предложена TNMB (tumor, node, metastasis, blood) — классификация кожных Т-клеточных лимфом, которая применима для определения стадий ГМ (табл. 1, 2) [8]. Данные о пятилетней выживаемости при грибовидном микозе/синдроме Сезари (ГМ/СС) в зависимости от стадии заболевания следующие: IA — 96 %, IB / IIA — 73 %, IIB / III — 44 %, IV — 27 %.
Фолликулотропный ГМ характеризуется наличием фолликулотропного и часто неэпидермотропного инфильтрата. Клинически заболевание может проявляться фолликулярными папулами, бляшками и иногда опухолями, локализующимися чаще всего на голове и шее и сопровождающимися алопецией. Гистологически выявляется плотный периаднексальный (перифолликулярный) инфильтрат, состоящий из малых и средних лимфоидных клеток с церебриформными ядрами. Эпидермотропизм может отсутствовать. Волосяные фолликулы часто кистозно расширены, возможна муцинозная дегенерация фолликулярного эпителия.
Синдром Сезари характеризуется триадой признаков: эритродермия, лимфаденопатия и наличие опухолевых Т-лимфоцитов (клеток Сезари) в коже и периферической крови [9].
Клинически синдром Сезари характеризуется развитием эритродермии с генерализованным зудом кожного покрова, увеличением периферических лимфатических узлов, наличием ладонно-подошвенного гиперкератоза и ониходистрофии. Интернациональным обществом по кожным лимфомам были предложены следующие критерии для диагностики синдрома Сезари: абсолютное количество клеток Сезари в периферической крови не меньше 1000 в мм 3 , увеличение соотношения CD4/CD8 лимфоцитов более чем в 10 раз и потеря Т-клеточных антигенов (CD2, CD3, CD4, CD5), подтверждение Т-клеточной клональности в периферической крови молекулярными или цитогенетическими исследованиями [10].
Гистологические изменения при синдроме Сезари сходны с таковыми при ГМ, однако эпидермотропизм может быть менее выражен.
Терапия ГМ/СС также зависит от стадии заболевания. На ранних стадиях, при локальных изменениях кожи, эффективна РUVA-терапия, которая позволяет получить полную ремиссию в подавляющем большинстве случаев [11–13]. При I и II стадиях PUVA-терапия может быть использована в комбинации с интерфероном α [14–16]. Применение агрессивных методов лечения (химио- и лучевая терапия) на ранних стадиях не меняет прогноз и, следовательно, неоправдано [17]. При распространенном поражении локальная лучевая терапия, тотальное облучение кожи (ТОК), экстракорпоральный фотоферез позволяют контролировать течение болезни, но имеют ограниченную доступность [18–21]. Когда заболевание становится резистентным к указанным методам лечения, используется комбинированная химиотерапия, однако вне зависимости от ее варианта продолжительность эффекта обычно не превышает 1 года [22–26]. В последнее время в лечении ГМ/СС все большее распространение получает применение биологических препаратов, механизм действия которых основан на специфическом связывании с различными антигенами на мембране опухолевых клеток. К ним относятся интегрированный протеин — Ontak и анти-CD52-моноклональное антитело — алемтузумаб (Кэмпас). Ontak является конъюгатом токсина дифтерии с интерлейкином-2, который после связывания с рецептором к интерлейкину-2 (СD25) подвергается эндоцитозу с последующим высвобождением внутри клетки дифтерийного токсина. Результатом этого процесса является нарушение синтеза белка и в конечном итоге апоптоз Т-лимфоцитов [27]. Кэмпас представляет собой гуманизированное IgG1-моноклональное антитело, специфически связывающееся с CD52-антигеном. Эффекторный механизм Кэмпаса изучен не до конца, он, вероятно, основан на антителозависимой клеточной цитотоксичности [28, 29], комплемент-обусловленном клеточном лизисе [30, 31] и апоптозе [32]. Опухолевые Т-лимфоциты экспрессируют на своей поверхности большое количество молекул CD52 (около 500 000 молекул на лимфоцит), и интенсивность экспрессии CD52 напрямую коррелирует с клиническим эффектом [33, 34]. Основанием для использования Кэмпаса при ГМ является его успешное применение при других Т-клеточных опухолях, например, при Т-клеточном пролимфоцитарном лейкозе [35]. По данным одного из наиболее крупных исследований по использованию Кэмпаса в терапии ГМ/СС, в которое было включено 22 больных со II–IV стадиями заболевания, ранее получавших другие виды лечения, общий ответ на терапию составил 55 %, полная ремиссия достигнута в 32 % случаев. Если предшествующее лечение включало в себя не более двух режимов терапии, общий ответ составлял 80 %. Медиана выживаемости без прогрессии для 12 больных, ответивших на лечение, составила 12 мес [36]. Клиническая картина до и после терапии ГМ препаратом Кэмпас представлена на рис. 1–4.
 |
| Рис. 1. Разрушение зоны эпидермально-дермального стыка. Распространение инфильтрата в эпидермис с образованием абсцессов Потрие. Рис. 2. Единичные лимфоидные клетки среди эпителиоцитов базального слоя эпидермиса (после терапии алемтузумабом) |
Первичные CD30 + -лимфопропролиферативные заболевания кожи занимают второе место по частоте возникновения среди всех Т-клеточных лимфом кожи. Они включают анапластическую крупноклеточную лимфому кожи и лимфоматоидный папулез. Их объединяющим признаком является экспрессия опухолевыми клетками CD30 — рецептора, принадлежащего к группе рецепторов фактора некроза опухолей.
Первичная анапластическая лимфома кожи чаще развивается у лиц мужского пола, в основном в возрасте старше 60 лет. Клинически определяются обычно один или несколько узлов (в том числе подкожных), имеющих тенденцию к изъязвлению. Наиболее частая локализация высыпаний — верхние и нижние конечности. Гистологическая картина представлена диффузными инфильтратами в дерме и подкожно-жировой клетчатке, состоящими из клеток с анапластической или иммунобластной морфологией. Эпидермотропизм не характерен. Опухолевые клетки экспрессируют CD30, а также один или несколько пан-Т-клеточных антигенов (CD2, CD3, CD4, CD5). Экспрессия антигена эпителиальных мембран (EMA) и ALK-протеина обнаруживается крайне редко, что отличает первичную кожную анапластическую лимфому от нодальной анапластической лимфомы с поражением кожи [37].
Первичная анапластическая лимфома кожи имеет, как правило, доброкачественное течение. Пятилетняя выживаемость составляет около 90 %. Лучевая терапия или хирургическое удаление применяются в случае единичных очагов. У больных с множественными высыпаниями проводится лечение малыми дозами метотрексата (5–20 мг в неделю) или лучевая терапия. Только быстропрогрессирующее течение заболевания с внекожным распространением требует назначения полихимиотерапии.
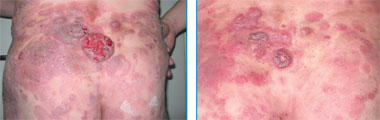 |
| Рис. 3. Множественные пятна, бляшки и опухолевые образования с элементами изъязвления на коже (до начала лечения). Рис. 4. Вид после терапии алемтузумабом |
Лимфоматоидный папулез — хроническое лимфопролиферативное заболевание кожи, характеризующееся рецидивирующим течением с повторными высыпаниями самопроизвольно разрешающихся папулезных элементов с гистологическими признаками CD30 + -лимфомы. Выделяют три гистологических варианта заболевания: тип А, тип В и тип С.
Тип А характеризуется кожным инфильтратом состоящим из крупных атипичных клеток, напоминающих клетки Рида–Штернберга.
Тип В гистологически напоминает картину ГМ. Анапластические клетки встречаются в малом количестве или отсутствуют.
Тип С характеризуется мономорфным инфильтратом или наличием крупных кластеров CD30 + Т-лимфоцитов. Наиболее эффективным методом лечения является назначение малых доз метотрексата, особенно в случае распространенных высыпаний и частых обострений [3].
Таким образом, диагностика и лечение первичных Т-клеточных кожных лимфом требуют учета многих факторов и использования широкого спектра современных методов обследования (гистологических, иммунологических, молекулярно-генетических), позволяющих правильно определить прогноз и оптимизировать терапию.
По вопросам литературы обращайтесь в редакцию.
В. А. Доронин
ЦКБ № 2 им. Н. А. Семашко, Москва
а) Определение. Грибовидный микоз является самым частым вариантом ТКЛК, обычно возникающим в возрасте 55-60 лет с большей предрасположенностью женщин 2:1.
б) Клиника грибовидного микоза:
1. Кожные симптомы. Клинически грибовидный микоз разделяется на стадии пятен, бляшек и опухолей, но у пациентов одновременно может присутствовать более одного типа поражений. На ранней стадии пятен грибовидного микоза определяются единичные или множественные эритематозные, чешуйчатые и обширные пятна, различные по размеру и обычно хорошо отграниченные.
Цвет поражений может варьировать от оранжевого до темного фиолетово-красного. Распространение обычно охватывает места, закрытые от солнца, с областями опрелостей, возникающими на ранней стадии заболевания. В областях поражения может возникать сильный зуд, но они могут быть и бессимптомными, иногда транзиторными, спонтанно исчезая без рубцевания. Диагноз на данной стадии затруднителен.
Пятна могут существовать месяцы или годы, прогрессируя до стадии бляшек, которые могут возникать de novo. Пятна четко отграничены, чешуйчатые, приподнятые, от темно-красного до фиолетового цвета, иногда уплотненные. Поражения на этой стадии могут спонтанно регрессировать или сливаться в обширные области с округлыми, изогнутыми или ползучими границами, могут обесцвечиваться в центральной области с оставшейся активностью по периферии. Иногда присутствует пурпурная гиперпигментация или гипопигментация и пойкилодермия.
Опухоли могут возникать на любом участке тела, но предпочтительно на лице и в складках тела: в подмышечных впадинах, в паху, локтевых ямках и в складках под грудью у женщин. Поражения могут возникать на месте предсуществующих бляшек или пятен ТКЛК, что указывает на развитие фазы вертикального роста. На этой стадии неопластические клетки более сходны по поведению со злокачественными, с вертикальным распространением, которое ведет к появлению разрастающихся дермальных узлов.
Появление de novo указывает на метастатическое распространение злокачественного клона Т-клеток. Узелки имеют красно-коричневый или пурпурно-красный цвет, мягкую поверхность, часто изъязвлены и могут вторично инфицироваться. Скорость роста различна. Пациенты с опухолями склонны к более агрессивной форме заболевания, чем пациенты на стадии пятен и бляшек.
2. Другие симптомы. Пациенты могут жаловаться на лихорадку, озноб, потерю веса, слабость, бессонницу вторично вследствие надоедающего зуда, а также плохое поддержание температуры тела. Могут присоединиться гиперкератоз, шелушение и растрескивание ладоней и стоп, алопеция, выворот века, дистрофия ногтей и отек лодыжек с блестящей и истонченной кожей. Эти изменения ведут к болезненности при ходьбе и значительных сложностях в ручной мелкой работе. Такие пациенты значительно ослаблены кожными проявлениями этого летального заболевания.
Зуд часто интенсивный, приводящий к расчесыванию, экссудации и вторичному инфицированию, что может доминировать в клинической картине.
б) Дифференциальная диагностика. Дифференциальный диагноз грибовидного микоза представлен в блоке ниже.

в) Гистология. В пятнах, бляшках и на стадии эритродермии присутствуют эпидермотропные пучковые инфильтраты неопластических Т лимфоцитов со складчатым сегментированным ядром, проникающие в верхний слой дермы с экзоцитозом и образованием внутриэпидермальных микроабсцессов Потрье. На опухолевой стадии в дерме обнаруживаются узловые инфильтраты, а эпидермальный компонент выражен меньше. Злокачественные клетки имеют фенотип зрелых периферических Т-клеток (CD4+).
При грибовидном микозе может возникать частичная потеря пан-Т-клеточных антигенов, таких как CD7 и CD3, ненатогномоничная для данного заболевания. При анализе генов Т-клеточного рецептора часто выявляется клональная реаранжировка, как показывают ПЦР и метод Саузерн- блота.
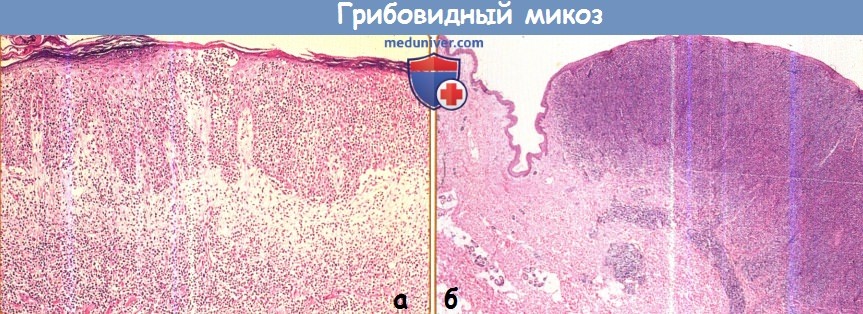
а - Плотный мононуклеарный инфильтрат, распространяющийся от сосочкового слоя дермы в эпидермис.
Эпидермис полностью пронизан этими клетками, формирующими абсцесс Потрье.
б - Опухоль при Т-клеточной лимфоме кожи на малом увеличении. Плотный инфильтрат проникает глубоко в дерму. 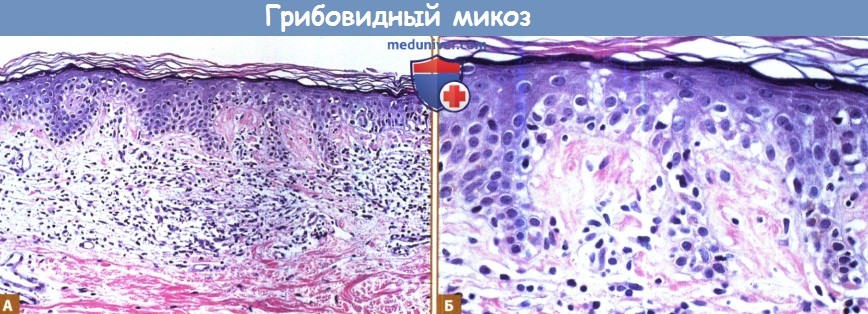
Стадия пятен Т-клеточной лимфомы кожи:
А. Единичные атипичные мононуклеарные клетки в эпидермисе с редкими поверхностными периваскулярными инфильтратами в сосочковом слое дермы (гематоксилин/эозин, окрашенный срез).
Б. Атипичные клетки эпидермиса полученные с того же среза при высоком разрешении.
д) Варианы грибовидного микоза (ГМ):
1. Фолликулотромный грибовидный микоз (ГМ). Фолликулотропный ГМ представляет отдельный вариант ГМ, характеризующийся преимущественным поражением Т-клеточными инфильтратами головы и шеи, со слизистой дегенерацией волосяных фолликулов или без нее. Ранее этот вариант назывался фолликулярный муциноз или слизистая аллопеция. Фолликулотропный ГМ возникает, в основном, у взрослых, но может изредка поражать детей и подростков. У пациентов могут присутствовать группы фолликулярных папул, акнеподобные поражения, уплотненные бляшки и иногда опухоли, обычно расположенные на голове и шее.
Часто возникают алопеция, зуд и вторичные бактериальные инфекции. Характерным явлением служат инфильтрированные бляшки на бровях с одновременной алопецией. Зуд часто тяжелый.
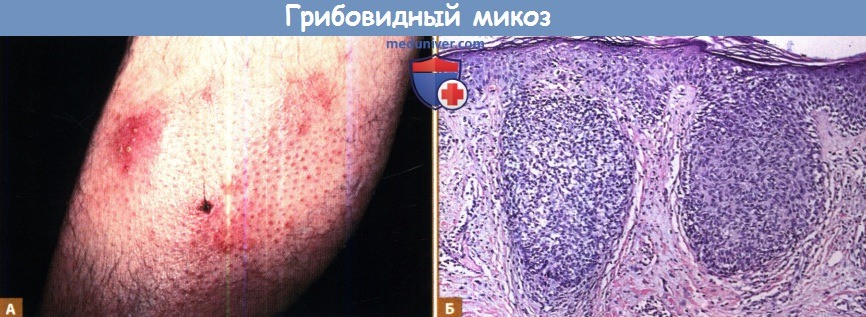
А. Фолликулотропный грибовидный микоз. Обратите внимание на локализацию в фолликулах, ведущую к потере волос.
Б. Внешняя оболочка корня волосяного фолликула разрушена Т-клетками и отложениями муцина, что привело к возникновению небольших кистозных полостей.
2. Гипопигментированный грибовидный микоз (ГМ). У пациентов с темной кожей развивается гипопигментированный ГМ, вариант стадии пятен. У темнокожих лиц это самое частое проявление заболевания. Пациенты отвечают на лечение восстановлением пигментации, а рецидив снова часто вызывает гипопигментацию.
3. Педжетоидрый ретикулез. Ретикулоз Педжета (болезнь Ворингера-Колоппа) является отдельным вариантом ГМ, характеризующимся локализованными пятнами и бляшками с внутриэпидермальной пролиферацией неопластических клеток. Пациенты поступают с единичным псориазоподобным или гиперкератозным медленно прогрессирующим пятном или бляшкой, обычно на конечностях. В отличие от классического ГМ, внекожная диссеминация не наблюдается. Раньше термин ПР также включал в себя диссеминированные формы (тип Кетрона-Гудмана).
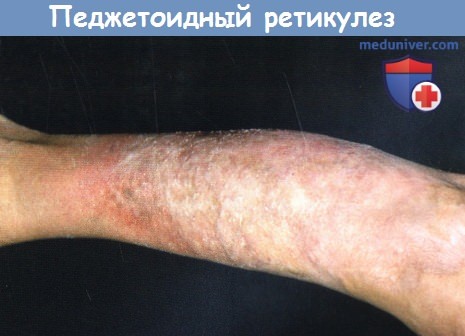
Педжетоидный ретикулез.
На голени пациента находится гиперкератозное пятно.
4. Синдром гранулематозной вялой кожи. Редкий подтип ГМ, характеризующийся медленно развивающимися участками провисающей кожи на крупных складках, преимущественно в подмышечной области и мошонке. При световой микроскопии выявляются плотные гранулематозные инфильтраты в дерме. Кроме мелких атипичных клеток с сегментированным ядром, вокруг некробиотической области в пределах разрушения эластической ткани встречаются макрофаги и многочисленные многоядерные гигантские Т-клетки. Неопластические клетки экспрессируют фенотип CD3 + CD4 + CD8 - .

Синдром гранулематозной вялой кожи. Обратите внимание на складки кожи вследствие вторичного эластолиза. 
- Рекомендуем далее ознакомиться со статьей "Кожа при синдроме Сезари (СС)"
Редактор: Искандер Милевски. Дата публикации: 3.1.2019
Содержание
Дерматология в России
- Сайт зарегистрирован как СМИ, электронное периодическое издание на русском и английском языке, ISSN 2077-3544
- Основатель и главный редактор проекта - проф. А.Ю. Сергеев
- Посещаемость сайта свыше 1500 человек в сутки (статистика). Среди более 15000 наших врачей-подписчиков 223 доктора и 1229 кандидатов медицинских наук, заведующие кафедрами, директора институтов и научных центров, руководители здравоохранения. Издание распространяется бесплатно, для использования профессиональных и интерактивных материалов необходима регистрация
- Дерматология в России выходит и обновляется практически ежедневно. Сайт предоставляет возможности коммуникации для практикующих врачей, ученых, преподавателей, представителей общественных организаций и фармацевтической индустрии
Обзор лечения грибовидного микоза и синдрома Сезари: подход, основанный на стадиях заболевания
Грибовидный микоз (ГМ) и синдром Сезари ( S é zary ) (СС) являются наиболее распространенными подтипами кожных Т-клеточных лимфом. У большинства больных они являются вялотекущими и неизлечимыми заболеваниями. Поэтому целью лечения должно быть достижение оптимальной выгоды при минимизации токсичности, насколько это возможно. Для достижения этой цели необходимо проводить лечение с учетом стадии заболевания. Лечение на ранней стадии ГМ ( IA - IIA ), включает в себя терапевтические средства, направленные на кожу и включает топические кортикостероиды, фототерапию, топическую химиотерапию, топические ретиноиды и лучевую терапию.
Для резистентных случаев ГМ на ранней стадии или на более поздних стадиях ГМ должна рассматриваться системная терапия в т.ч. и альфа-интерферон, оральные ретиноиды, в т.ч. бексаротен ( bexarotine ) и в последнее время ацитретин, ингибиторы гистонов деацетилазы ( HDACi ), токсин denileukin diftitox и химиотерапевтические препараты. В некоторых ситуациях может быть рассмотрено сочетание лекарственного лечения, чтобы получить синергетический эффект, позволяющий снизить дозы препаратов. Химиотерапия должна всегда назначаться в случаях, когда другие системные препараты были опробованы или противопоказаны. Новые препараты следует использовать в случаях неэффективности традиционных системных препаратов.
Ключевые слова: кожная T -клеточная лимфома, грибовидный микоз, синдром Сезари
Первичные кожные лимфомы состоят из Т-клеточных (75%) и реже B-клеточных лимфом и составляют 2% всех лимфом с ежегодной заболеваемостью 0,3 до 1 на 100 000. Существуют различные типы кожных Т-клеточных лимфом (КТКЛ). До относительно недавнего времени было 2 классификации КТКЛ - Всемирной организации здравоохранения (ВОЗ) и европейской организации исследования и лечения рака (EORTC) Последняя характеризуется делением на агрессивные или вялотекущие, основанные на клинико-патологических критериях. В 2005 году эти 2 классификации были объединены ( Таблица 1 ).
WHO-EORTC классификации кожной T-клеточной лимфомы
Подтипы грибовидного микоза
Гранулематозная дряблая кожа
Первичные кожные CD 30 лимфопролиферативные заболевания
Первичная кожная анапластическая крупноклеточная лимфома
Подкожная паникулитоподобная Т-клеточная лимфома
ГМ и его лейкозный вариант СС являются наиболее распространенными формами КТКЛ. Ежегодная частота КТКЛ, как сообщается, растет и в настоящее время оценивается на уровне 9,6 случаев на 1 млн человеко-лет. Долгосрочное выживание большинства пациентов приводит к гораздо более высокой общей распространенности. Продлению жизни способствуют методы, воздействующие на кожу, такие как фототерапия, топические препараты, и лучевая терапия; растет арсенал системных препаратов, начиная от ретиноидов и до препаратов для химиотерапии. Появился новый метод аллогенной трансплантации стволовых клеток. Однако нет достаточного количества рандомизированных исследований, чтобы рекомендовать предпочтительную стратегию лечения для ГМ/СС и не существует общепринятого стандарта лечения. В данной статье представлен краткий обзор этапного лечения ГМ/СС.
Этиология ГМ остается неизвестной. Однако различные теории предполагают профессиональные факторы или воздействие факторов окружающей среды, цитокинов, онкогенов, других видов хронической антигенной стимуляции или вирусное воздействие.
Варианты грибовидного микоза
Классический тип ГМ подразделяется на 4 подтипа: эритематозный, бляшечный, опухолевый и эритродермию или СС. Из них наиболее распространенным является эритематозный подтип ГМ, первоначально описанный Алиберт и представленный сильно зудящими, эритематозными пятнами, участками атрофии и телеангиэктазиями. Однако многие клинические и гистологические варианты имеют необычные или уникальные клинические проявления, такие как эритродермический, фолликулярный, сиринготрофический, буллезный/везикулярный, гранулематозный, гиперпигментированный, пойкилодермический, гиперкератотический, папилломатозный, ихтиозиформный, ладонно-подошвенный, односторонний, пигментированно-пурпурозный, импетигинозный, педжетоидный ретикулез и внекожный. СС-это отдельная (лейкозная) форма КТКЛ, при которой значительную роль играют клетки крови и развивается эритродермия и лимфаденопатия. Часто при СС наблюдаются дополнительные клинические данные в виде поражения роговицы, дистрофии ногтей, алопеции, эктропиона и отека кожи (особенно на ногах). Эти больные часто испытывают мучительный зуд, который может быть наиболее значимым симптомом, снижающим качество жизни, и поэтому для пациента наибольшее значение имеют процедуры, направленные на уменьшение зуда, даже без внешнего клинического улучшения.
Диагностика и прогноз
Эритема и бляшки возникают в I стадии, которая разделена на IA ( 10% поверхности тела). Наличие клинически выраженной лимфаденопатии без патологической узловой инфильтрации представляет стадию IIA, кожные опухоли характеризуют стадии IIB, генерализованная эритродермия характеризует стадию III, вовлечение в процесс лимфатических узлов характерно для стадии IVA, висцеральные поражения характерны для стадии IVB. Стадии IA, IB и IIA считаются ранними стадиями заболевания, а стадии IIB (опухоль), III (эритродермия) и IV (вовлечение лимфоузлов узлов с или без вовлечения внутренних органов) -поздними стадиями заболевания.
Хотя ГМ/СС обычно считаются неизлечимыми, важно признать, что у большинства пациентов наблюдаются вялотекущие формы заболевания и они могут жить много лет. Действительно, подсчитано, что 65% - 85% пациентов с ГМ имеют стадии IA или IB заболевания. Наиболее важным фактором в планировании и определении прогноза является стадия заболевания. Действительно, большинство пациентов с ранними стадиями заболевания (стадии IA, IB и IIA) не переходят в поздние стадии болезни, и пациенты с изолированными эритемами или бляшками (T1-T2) имеют среднюю выживаемость более 12 лет. Больные с поздними стадиями заболевания (стадии IIB, III и IVA) с опухолями, эритродермиями, и вовлечением лимфатических узлов илиизменениями в крови, но без вовлечения висцеральных органов, имеют в среднем выживаемость 5 лет. Следует отметить, что больные с опухолями (T3) живут меньше, чем с эритродермией (T4). Больные с висцеральными поражениями встречаются редко (стадия IVB) и живут в среднем всего 2,5 года или меньше.
В ранней стадии ГМ есть некоторая прогностическая неоднородность. Действительно, между ранней и поздней стадиями заболевания имеются группы “промежуточного риска”. Это включает в себя пациентов со стадией IIA/IB фолликулотропного варианта ГМ и пациентов с очень толстыми бляшками. Относительно худшие результаты в этих группах связаны со снижением реактивности на кожно-направленную терапию. Пациенты с поздней стадией болезни, со стадией IIB заболевания с несколькими опухолевыми узлами (увеличением опухолевой массы) и крупноклеточной трансформацией ГМ имеют значительно худший прогноз. Низкая численность CD8+ Т-клеток в дермальном инфильтрате и/или крови также были независимо связаны со снижением выживаемости.
Для пациентов с клинически очень ограниченной стадией болезни с пятнами на коже и/или бляшками без пальпируемых лимфатических узлов, исследование обычно не требуется. Иногда у пациентов выявляется региональная лимфаденопатия, которая может отражать дерматопатические изменения в узле, а не участие узлов в ГМ. Таким образом, не всегда необходима биопсия у каждого пациента с умеренно увеличенными узлами. В общем, рекомендуется биопсия узлов размером более 1,5 см, как имеющих существенное прогностическое влияние. Относительная нерешительность в выполнении биопсии узла связана с высокой частотой колонизации кожи патогенными микроорганизмами у пациентов с ГМ/СС, что увеличивает риск инфекции после операции.
Выбор подходящего лечения основывается прежде всего на стадии, в зависимости от TMNB классификации. Однако, другие прогностические переменные, такие как крупноклеточная трансформация, также должны быть рассмотрены. Дополнительные факторы, учитываемые при подборе лечения включают возраст пациента, общее состояние здоровья, остроту и тяжесть сопутствующих симптомов (напр., зуд, опухоли, изъязвления, скорость реакции, время и продолжительность ответа на лечение, данные о связанных с лечением побочных эффектах и доступность или затраты-выгоды процедур. Как правило, лечение делится на кожно-направленную терапию [SDT], системную терапию и комбинированную терапию.
Лечение грибовидного микоза/синдрома Сезари. *
1. Кожно-направленная терапия
2. Системнаяная терапия
1.1. С ограниченным/локализованным поражением кожи
· Топическая химиотерапия (мехлорэтамин [азота горчица], кармустин)
· Местное облучение (12-36 гр)
· Топические ретиноиды (бексаротен, тазаротен)
· Фототерапия (UVB, NBUVB для пятен/тонких бляшек; ПУВА-для более толстых бляшек
2.1. Категория (SYST-CAT)
· Ретиноиды (бексаротен, полностью транс-ретиноевая кислота, изотретиноин [13-цисретиноевая кислота], ацитретин)
· Интерфероны (ИФН-альфа, ИФН-гамма)
· HDAC-ингибиторы (см. текущие исследования, ромидепсин)
· Денилейкин дифтитокс (Denileukin diftitox)
· Метотрексат (100 мг в неделю)
1.2. Для обобщенного поражениея кожи
Топическая химиотерапия (мехлорэтамин [азота горчица], кармустин)
Фототерапия (UVB, NB UVB, для пятен/тонких бляшек; ПУВА-для более толстых бляшек)
Итого кожи электронно-лучевой терапии (30-36 гр) d (зарезервировано для людей с тяжелой или генерализованные кожные проявления толстые бляшки или опухоли, болезни или плохой ответ на другие терапии)
2.2. Категория B (SYST-CAT B)
· Терапия первой линии Липосомальныйдоксорубицин
· Терапия второй линии
· Метотрексат (>100 мг в неделю)
· Низкие дозы пралатрексата
Лечение ранней стадии (IA-IIA) грибовидного микоза
Как уже упоминалось, большинство пациентов имеют заболевания на ранней стадии. Терапия не влияет на выживание, а неагрессивный подход к терапии является оправданным при лечении, направленном на улучшение симптомов и косметических дефектов с ограничением при этом токсичности. Пациенты со стадией IA заболевания могут жить достаточно долго. “Выжидательная тактика” может быть вполне законным вариантом у данных пациентов, при условии, что она включает в себя тщательный мониторинг. Учитывая, что в начальной стадии лечения часто имеются множественные участки кожи, в первую очередь проводится кожно-направленная терапия. Основным выбором для кожно-направленной терапии являются топические кортикостероиды, ПУВА-терапия или UVB. Действительно, для пациентов с ограниченной эритематозной формой заболевания топические стероиды часто позволяют контролировать болезнь в течение многих лет, и часто это единственный вид терапии, необходимой для таких пациентов. Класс I (сильнодействующие) топические кортикостероиды, такие как бетаметазон дипропионат 0,05% или мометазона фуроат 0,1%, являются наиболее эффективными. Пациенты со стадией T1 заболевания при лечении топическими стероидами имеют полный ответ примерно в 60% - 65% и частичный ответ в 30%. Пациенты со стадией T2 (генерализованные эритематозные высыпания с вовлечением >10% поверхности кожи) имеют полный ответ в 25% и частичный ответ в 57%. Внутриочаговое введение кортикостероидов может быть эффективным в лечении утолщенных поражений ГМ, таких как бляшки или опухоли. Для более распространенных поражений кожи рекомендуются ПУВА или UVB. Полный эффект при ПУВА-терапии у больных в начальной стадии болезни достигается в 58%- 83% и частичный эффект- у 95%. Кроме того, ремиссия часто длительная в среднем до 43 месяцев. Поддерживающее лечение в виде недельных или двухнедельных курсов терапии может быть эффективным в продлении ремиссии. ПУВА-терапия обычно хорошо переносится; однако, острые побочные эффекты включают тошноту (от орального псоралена) или фотосенсибилизацию. Долгосрочные побочные эффекты - ускоренное актиническое повреждение и повышенный риск злокачественных новообразований кожи, включая плоскоклеточный рак и меланому.
Рекомендации для лечения грибовидного микоза IA, IB, IIA стадий
UVB также эффективен для ГМ, особенно при наличии эритематозных очагов и малой их толщине. Узкополосная фототерапия (311 нм) также эффективна при ГМ, хотя ремиссия после нее может быть короче. Преимущество UVB над ПУВА в том, что она более доступна и позволяет избежать побочных эффектов. Недостатком UVB является меньшая скорость реакции, более короткая ремиссия и меньшая эффективность, чем у ПУВА при более толстых поражениях. Добиться лучшего ответа удается при сочетании ПУВА с интерфероном Альфа-2b. или ретиноидами, такими как ацитретин. ПУВА-терапия используется также в качестве " спасительной " или поддерживающей терапии после тотальной терапии кожи электронным пучком (TSEB). Для толстых бляшек, особенно при локализованных поражениях, эффективна радиотерапия, т.к. заболевание является особо радиочувствительным. К другим вариантам терапии первой линии относится топическая химиотерапия с использованием мехлорэтамина или кармустина. Однако, использование этих средств нецелесообразно при обширных поражениях. К побочным реакциям мехлорэтамина относятся аллергический контактный дерматит, ксероз кожи, гиперпигментация, телеангиэктазии, а также повышенный риск плоскоклеточного и базально-клеточного рака кожи. При применении водного транспортного средства гиперчувствительность к мехлорэтамину возникает в 40%. Гиперчувствительность наблюдается реже при использовании препарата в форме мази. Ко второй линии терапии на ранних стадиях ГМ, применяемой при неэффективности топических кортикостероидов, относятся ретиноиды, такие как бексаротен и ацитретин, эффективность которого была продемонстрирована совсем недавно в ретроспективном исследовании. Ацитретин хорошо переносится и потенциально эффективен на ранних стадиях КТКЛ с сопоставимым результатом с другими пероральными агентами, утвержденными для данного заболевания, такими как ИФН-Альфа, низкодозированный оральный метотрексат, ингибиторы гистонов деацетилазы (HDACi), или денилейкин дифтитокс (denileukin diftitox). Радиотерапия-это высокоэффективная терапия в ГМ/СС и может быть использована как на ранних, так и поздних стадиях заболевания. Частичная регрессия заболевания может наблюдаться и при разовых дозах, как низкий, как и 1.0 гр. Комбинации кожно-направленной терапии с системной терапией показаны при неэффективности монотерапии, при тяжелых кожных симптомах или при наличии других неблагоприятных прогностических факторов. У пациентов с поздней клинической стадией (≥ IIB) кожно-направленная терапия используется в комбинации с системными методами или в качестве поддерживающей терапии. Излучение используется последовательнов сочетании с другими методами лечения: ПУВА -, UVB, ретиноиды и топическая или системная химиотерапия. Иногда лечение может быть назначено одновременно, но при обработке больших участков кожи дозы облучения должны быть модифицированы чтобы свести к минимуму риск покраснения или шелушения.
Лечение поздних стадий (IIB-IVB) грибовидного микоза
Лечение поздней стадии болезни, или обострений ранней стадии болезни является более проблематичным и всегда требует мультидисциплинарного подхода. Прежде чем начинать химиотерапию рекомендуется лечение ИФН-альфа, бексаротеном или денилейкин дифтитоксом.
Трансплантация стволовых клеток
Делать выводы об эффективности метода трансплантации стволовых клеток сложно, поскольку количество пациентов с ГМ/СС, прошедших лечение этим методом очень мало. Аллогенная трансплантация лучше подходит молодым пациентам с развитой стадией болезни, не реагирующим на лечение IFN-альфа, бексаротеном или денилейкин дифититоксом. Результаты аутологичной трансплантации стволовых клеток не были особенно перспективными. Ясно, что для этой группы пациентов необходимы большие исследования.
Новые агенты в ходе клинических исследований
Новые препараты, которые исследуются в рамках клинических испытаний, должны быть рассмотрены для клинических испытаний в качестве альтернативных, когда другие препараты не эффективны.
Важным шагом в лечении ГМ является определение клинической стадии, что требует хорошей клинико-патологической оценки. Для этого необходимо регулярное наблюдение и повторные биопсии. Лечение должно быть индивидуальным в зависимости от стадии заболевания и состояния здоровья пациента, чтобы избежать излишне агрессивной терапии, включая химиотерапию. Применение новых лекарственных средств целесообразно в случаях, когда известные системные препараты не эффективны.
Читайте также: