Аномалия Чедиака-Штейнбринка-Хигаши (Chediak-Steinbrinnk-Higashi) - причины, клиника
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Синдром Чедиака-Хигаси — очень редкое наследственное заболевание, характеризующееся периодическими бактериальными респираторными и другими инфекциями и отсутствием пигмента в волосах, глазах и коже (альбинизм).
У людей с синдромом Чедиака-Хигаси обычно бледная кожа, светлые или белые волосы и розовые или бледно-голубые глаза.
Врачи исследуют образец крови для исключения аномалий и выполняют генетические тесты, чтобы подтвердить диагноз.
Лечение включает антибиотики для профилактики инфекций, другие препараты для улучшения функции иммунной системы и, по возможности, трансплантацию стволовых клеток.
Люди с синдромом Чедиака-Хигаси более восприимчивы к инфекциям, поскольку фагоциты (типы лейкоцитов Компоненты иммунной системы Иммунная система защищает организм от чужеродных или опасных элементов. Такие элементы включают: микроорганизмы (обычно называемые микробами, такие, как бактерии, вирусы и грибки); паразиты. Прочитайте дополнительные сведения , включая нейтрофилы, эозинофилы, моноциты и макрофаги), не функционирует нормально. Фагоциты — это клетки, которые поглощают и уничтожают микроорганизмы.
Симптомы синдрома Чедиака-Хигаси
При синдроме Чедиака-Хигаси пигмент меланин образуется в очень малых количествах либо не образуется совсем (это называется альбинизм Альбинизм Альбинизм — это редкое наследственное заболевание, при котором образуется мало кожного пигмента меланина или он не образуется вообще. Поражаются кожа, волосы и глаза, а иногда только глаза. Прочитайте дополнительные сведенияЗаболевание может также вызывать проблемы со зрением. Например, острота зрения может быть снижена, а чувствительность глаз к свету (фоточувствительность) повышена. Часто встречается нистагм, который вызывает нечеткость зрения. При нистагме глаза постоянно движутся: они быстро поворачиваются в одном направлении, затем медленно возвращаются в исходное положение.
Синдром Чедиака-Хигаси также часто приводит к язвам в полости рта, гингивиту и заболеванием периодонта.
Для лиц с синдромом Чедиака-Хигаси также характерны частые инфекции. Инфекции обычно поражают дыхательные пути, кожу и оболочки, выстилающие полость рта.
Приблизительно у 80% пациентов синдром Чедиака-Хигаси прогрессирует, вызывая лихорадку, желтуху Желтуха у взрослых При желтухе кожа и белки глаз желтеют. Желтуха появляется, когда в крови слишком много билирубина (желтый пигмент) — заболевание, называемое гипербилирубинемией. (См. также Общие сведения о. Прочитайте дополнительные сведенияСиндром Чедиака-Хигаси ( Синдром Бегеса-Чедьяка-Хигаши )
Синдром Чедиака-Хигаси - это одна из форм врожденного иммунодефицита, для которой характерен дефект фагоцитоза и глазокожный альбинизм. Заболевание имеет аутосомно-рецессивный механизм наследования и возникает при разнообразных типах мутаций LYSR/CHS1. Клинически патология проявляется обесцвечиванием волос и радужки глаз, рецидивирующими бактериальными и грибковыми инфекциями, гемофагоцитарным лимфогистиоцитозом. Программа диагностики включает миелограмму, клинические и иммунологические анализы крови, генетическое консультирование. Наиболее эффективным методом лечения признана трансплантация костного мозга.
МКБ-10



Общие сведения
Синдром Чедиака-Хигаси (СЧХ) - редкая форма генерализованной клеточной дисфункции, которая была описана в 1952 г. кубинским врачом Чедиаком и в 1954 г. японским педиатром Хигаси. Молекулярно-генетические основы патологии были детально изучены только спустя 34 года. В медицинской литературе описано около 500 случаев заболевания у детей младшего возраста. Несмотря на редкую встречаемость, синдром не теряет своей актуальности в практической генетике и педиатрии, поскольку он отличается неблагоприятным течением и требует усовершенствования методов лечения.

Причины
Синдром Чедиака-Хигаси вызван мутацией гена LYSR/CHS1, локализованного на длинном плече 1-й хромосомы. Он состоит из 53 экзонов и кодирует протеин, отвечающий за образование и транспорт клеточных органелл. Заболевание наследуется по аутосомно-рецессивному пути. Известно 63 варианта генетических мутаций, которые отличаются по тяжести патофизиологических нарушений. Наиболее благоприятными признаны миссенс-мутации, при которых СЧХ имеет мягкий фенотип.
Патогенез
Генетическая мутация LYST нарушает образование соответствующего белка и изменяет течение регулируемых им процессов. При синдроме образуются гигантские внутриклеточные гранулы, которые располагаются в лизосомах, цитолитических везикулах, меланосомах. Они нарушают функции клеток, прежде всего цитотоксических Т-лимфоцитов, которые отвечают за фагоцитоз чужеродных микроорганизмов. Также нарушается восстановление плазматических клеточных мембран.
Патогенетические изменения снижают активность фагоцитирующих лимфоцитов и натуральных киллеров. Неконтролируемое слияние лизосом и нарушение хемотаксиса - причины неспособности клеток к перевариванию фагоцитированных микроорганизмов. Патогены длительное время находятся внутри иммунных клеток, избегая токсического влияния антибиотиков. Помимо иммунной функции, нарушается транспорт меланина, с чем связаны проявления альбинизма.
При синдроме Чедиака-Хигаси в клетках крови и костного мозга возникают гигантские пероксидазоположительные гранулы, которые являются результатом объединения первичных и вторичных лизосом. Морфологически болезнь характеризуется лимфогистиоцитарной инфильтрацией паренхимы печени, лимфатических узлов, селезенки. Также происходят процессы эритрофагии.
Заболевание проявляется в первые годы жизни. Типичный внешний признак - парциальный альбинизм. Цвет волос варьирует от светло-желтого до седого, что зависит от типа мутации и этнической принадлежности. Радужка глаз имеет голубой или светло-серый оттенок. Пациенты чувствительны к свету: при длительном пребывании в ярко освещенном помещении глаза начинают слезиться и болеть. Фоточувствительность сопровождается снижением зрения, косоглазием.
Второй характерный синдром при СЧХ ‒ рецидивирующие бактериальные инфекции. Чаще всего патологии вызваны стрептококковыми и стафилококковыми возбудителями, при критическом снижении иммунной функции присоединяются оппортунистические инфекции. С раннего возраста дети страдают затяжными бронхитами, пневмониями, отитами, фурункулезом. На фоне антибиотикотерапии нередко развиваются грибковые инфекции - орофарингеальный кандидоз.
Для СЧХ характерна многообразная неврологическая симптоматика. У большинства детей развивается атаксия, сенсорные и моторные нарушения. Интенсивность клинических проявлений коррелирует с общей тяжестью заболевания и возрастом пациента. Вследствие прогрессирующей нейродегенерации возникают и усугубляются признаки умственной отсталости.
Осложнения
Неблагоприятным последствием синдрома является фаза «акселерации» - гемофагоцитарный лимфогистиоцитоз. Она возникает у 65-85% пациентов к 7-10 годам жизни, зачастую провоцируется вирусом Эпштейна-Барр (ВЭБ). Осложнение характеризуется воспалительной реакцией, неконтролируемой активацией макрофагов поражением внутренних органов. Клинически это проявляется лихорадкой, желтухой, панцитопенией и гепатоспленомегалией.
Несмотря на усовершенствование диагностики и медицинской помощи пациентам с синдромом Чедиака-Хигаси, уровень летальности остается высоким. Более 85% детей не доживают до возраста 10 лет. Среди основных причин смерти называют сепсис на фоне затяжных бактериальных инфекций, формирование злокачественных новообразований. При лечении методом ТГСК есть риск развития реакции «трансплантат против хозяина» с показателем смертности около 50%.
Диагностика
Заболевание имеет характерные внешние признаки, поэтому его удается заподозрить на приеме у педиатра. Помимо осмотра волос, глаз и кожных покровов, проводят стандартное физикальное обследование, изучают работу легких и сердца, выполняют пальпацию брюшной полости и определение размеров печени. Для подтверждения диагноза синдрома Чедиака-Хигаси требуется консультация генетика и дополнительные методы обследования, в числе которых:
- УЗИ органов брюшной полости. При сонографии определяется увеличение печени, поджелудочной железы и селезенки. При прогрессирующем заболевании наблюдаются диффузные изменения печеночной паренхимы, свободная жидкость в полости брюшины.
- Рентгенография ОГК. При рентгенологическом исследовании грудной клетки зачастую выявляются признаки пневмонии, гидроперикарда, плеврита. Для более детальной диагностики назначают МСКТ.
- Анализы крови. В гемограмме обнаруживают анемию, тромбоцитопению, нейтропению. Биохимическое исследование крови показывает повышение уровня триглицеридов и ферритина. По результатам коагулограммы определяют уменьшение концентрации фибриногена.
- Уточняющие исследования. В крови пациентов с СЧХ определяется низкая цитотоксическая активность NK-клеток, уровень растворимых CD25 более 2400 Ед/мл. Патогномоничный признак болезни в миелограмме - обнаружение пероксидазоположительных гранул в клетках костного мозга.
- Генетическая диагностика. Вследствие дороговизны и сложности проведения определение точного вида мутации LYSR/CHS1 при СЧХ назначается редко. Такая информация имеет клиническое значение для определения прогноза, составления программы генетического консультирования членов семьи.
Дифференциальная диагностика
При подозрении на болезнь Чедиака-Хигаси необходимо исключить другие варианты иммунодефицитов, сопровождающиеся альбинизмом и гемофагоцитозом, - синдромы Грисцелли и Германски-Пудлака. В сложных случаях дифференциальная диагностика проводится с семейной формой гемофагоцитарного лимфогистиоцитоза, изолированным глазокожным альбинизмом, синдромом Кросса-Мак-Кьюсика-Брина.
Лечение синдрома Чедиака-Хигаси
Консервативная терапия
Медикаментозные методы носят профилактический характер. Для предупреждения тяжелых инфекций проводят индивидуальные курсы антибактериальной и противогрибковой терапии, по показаниям назначают противовирусные препараты. Ежемесячно вводят внутривенные иммуноглобулины, препараты для стимуляции гранулоцитопоэза. Для профилактики фотоофтальмии пациентам рекомендуют носить затемняющее очки, избегать пребывания на ярком солнце.
Интенсивное лечение назначается в периоде «акселерации». Для коррекции воспалительного процесса применяются иммуносупрессоры и глюкокортикостероиды в высоких дозах. При сопутствующем поражении ЦНС препараты вводятся в спинномозговой канал или субарахноидальное пространство. Если осложнения вызваны ВЭБ-инфекцией, применяют моноклональные антитела. Правильно подобранная схема лечения позволяет достичь ремиссии у 75% пациентов в течение 2 месяцев.
Хирургическое лечение
Трансплантация гемопоэтических стволовых клеток (ТГСК) из костного мозга - наиболее перспективный вариант лечения, который повышает шансы на продление жизни при синдроме Чедиака-Хигаси. ТГСК рекомендуют выполнять до начала периода «акселерации»: в этом случае общая 5-летняя выживаемость составляет 62%. При позднем проведении пересадки костного мозга вероятность успеха составляет не более 42%.
Для уменьшения нагрузки на организм ребенка подготовку проводят в режиме кондиционирования со сниженной интенсивностью. Наилучшие результаты достигают при проведении трансплантации от HLA-совместимого родственного донора. В посттрансплантационном периоде продолжается иммуносупрессивная терапия, пациенты находятся в особых стерильных боксах для предупреждения развития инфекций.
Прогноз и профилактика
Пациенты с классической формой СЧХ живут не более 10 лет и умирают в фазе «акселерации». При проведении ранней костномозговой трансплантации качество жизни больного улучшается, процент выживаемости увеличивается, однако прогноз остается относительно неблагоприятным. Семейной паре, у которой родился ребенок с синдромом Чедиака-Хигаси, при планировании следующей беременности показана консультация генетика.
1. Синдром Чедиака-Хигаси (обзор литературы)/ Ю.А. Родина, В.Е. Матвеев, Д.Н. Балашов, М.Э. Дубровина, А.Ю. Щербина// Вопросы гематологии/онкологии и иммунопатологии в педиатрии. - 2016. - №15.
2. Гемофагоцитарный лимфогистиоцитоз/ М.А. Масчан, Г.А. Новичкова// Вопросы современной педиатрии. - 2009. - №3.
3. Chediak-Higashi syndrome/ J. Kaplan, I. De Domenico, and D. M. Ward// Curr. Opin. Hematol. - 2008. - №5.
Синдром Чедиака-Хигаси
Синдром Чедиака-Хигаси является редким аутосомно-рецессивным синдромом, характеризующимся нарушением лизиса фагоцитированных бактерий, в результате чего развиваются рецидивирующие бактериальные респираторные и прочие инфекции, также отмечается альбинизм кожи и глаз. Генетическое тестирование на LYST-мутацию может подтвердить диагноз. Лечение включает в себя профилактический прием антибиотиков, гамма-интерферон, и в некоторых случаях кортикостероиды Иногда трансплантация стволовых клеток имеет излечивающий эффект.
Симптомы и признаки синдрома Чедиака-Хигаси
Клинические проявления синдрома Чедиака-Хигаши включают глазокожный альбинизм Патофизиология Кожноглазной альбинизм - это наследуемый дефект образования меланина, вызывающий диффузную гипопигментацию кожи, волос и глаз. Глазной альбинизм поражает глаза, но, как правило, не кожу. Поражение. Прочитайте дополнительные сведенияУ 80% пациентов отмечается острое развитие заболевания с повышением температуры, желтухой, гепатоспленомегалией, лимфаденопатией, панцитопенией, геморрагическим диатезом и неврологическими изменениями. Острое развитие заболевания при синдроме Чедиака - Хигаси обычно фатально в течение 30 месяцев.
Диагностика синдрома Чедиака-Хигаси
Обычно наблюдается нейтропения, снижение естественной цитотоксичности клеток-киллеров, и гипергаммаглобулинемия. Исследуются мазки периферической крови на наличие гигантских гранул в нейтрофилах и других клетках; мазки костного мозга исследуются на наличие гигантских включений в клетках-предшественниках лейкоцитов.
Диагноз синдрома Чедиака-Хигаси может быть подтвержден генетическим исследованием на наличие мутаций LYST.
Поскольку это нарушение развивается крайне редко, нет необходимости осуществлять скрининг родственников, если клиническое подозрение не является высоким.
Поддерживающая терапия с использованием антибиотиков, гамма-интерферона, в некоторых случаях кортикостероидов
Трансплантация гемопоэтических стволовых клеток
Профилактическое применение антибиотиков может помочь предотвратить инфекции, применение гамма-интерферона может быть полезно для восстановления некоторых функций иммунной системы. Пульс-терапия кортикостероидами и спленэктомия иногда вызывают кратковременную ремиссию синдрома Чедиак-Хигаши.
Тем не менее, большинство пациентов с синдромом Чедиака-Хигаши возрастом до 7 лет умирают от инфекций, если не будет проведена трансплантация гемопоэтических стволовых клеток Трансплантация гемопоэтических стволовых клеток Трансплантация гемопоэтических стволовых клеток (ГСК) - быстроразвивающаяся технология, которая потенциально может позволить добиться излечения при злокачественных заболеваниях крови ( лейкемиях. Прочитайте дополнительные сведения . Может быть эффективна трансплантация костного мозга с идентичным нефракционированным главным комплексом гистосовместимости человека (НLA) после претрансплантационной циторедуктивной химиотерапии. Пятилетняя выживаемость после трансплантации составляет примерно 60%.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.

Синдром Чедиака-Хигаси — редкое аутосомно-рецессивное заболевание, которое характеризуется тяжелым врожденным иммунодефицитом по причине дефекта гена CHS1/LYST. Проявляется болезнь частыми бактериальными инфекциями, коагулопатией, нарушениями пигментации кожи, а также прогрессирующими неврологическими расстройствами [1].
Иммунологические нарушения выражаются в нейтропении и нехватке естественных киллеров (NK-клеток). Зачастую заболевание оказывается летальным — большая часть пациентов умирает в детстве. Заподозрить синдром Чедиака-Хигаси можно по тем же признакам, что и любой врожденный иммунодефицит, однако имеется и довольно специфичный критерий — гигантские внутриклеточные включения (или органеллы) в популяциях разных клеток и определенные дефекты в них [2].
Чтобы более наглядно описать происходящие процессы при данном заболевании, еще во второй половине прошлого столетия была предложена оригинальная модель «бежевой мыши». Вследствие мутации в определенном гене (в то время конкретная точка генома, разумеется, не была известна) пигментация экспериментальной мыши изменялась, и исследователи определили ее цвет как «бежевый» («beige»). Помимо цвета у таких животных имелся еще ряд аномалий в виде огромных гранул меланина в лимфоцитах, нейтрофилах и эозинофилах (рис. 1), также подобные включения наблюдались в ряде других клеток [3].
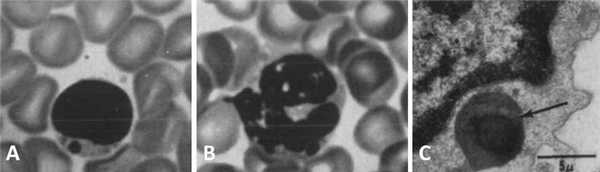
Рисунок 1 | Периферическая кровь бежевой мыши. Световая микроскопия: определяются гранулы в цитоплазме лимфоцита (А) и нейтрофила (В). Электронная микроскопия: объемная гранула с темным содержимым в цитоплазме лимфоцита. Относится к непостоянным аномальным включениям [3].
Позднее данный ген у мыши был идентифицирован и назван Lyst (кодирует регулятор лизосомального транспорта — lysosomal trafficking regulator), человеческий ген обозначают как LYST. Фермент, за экспрессию которого отвечает LYST, обеспечивает нормальный экзо- и эндоцитоз лизосомы; в случае мутации лизосома не может выполнять свои функции [4].
Сегодня этот ген настолько прочно ассоциируется с синдромом Чедиака-Хигаси, что в современной литературе его обозначают как CHS1/LYST(человеческий ген Chediak-Higashi syndrome-1 или LYST — оба варианта равнозначны) [1].
Роль регулятора лизосомального транспорта (LYST)
Данный белок кодируется достаточно объемным геном LYST, который содержит 55 экзонов и расположен в локусе 1q43. Большая длина нуклеотидной последовательности этого гена — фактор неоднозначный: с одной стороны, это существенно затрудняет диагностику, с другой — большое количество мутаций вообще никак не влияет на жизнедеятельность человека и не вызывает синдром Чедиака-Хигаси [5].
Ген кодирует одноименный большой каталитический белок — регулятор лизосомального транспорта (425 kDa, 3801 аминокислотное основание). Протеин относится к семейству BEACH — (Beige and Chediak-Higashi) — и, по-видимому, стал первым его представителем [4]. Белки данного семейства имеют три общих С-концевых домена: PH (Pleckstrin-homology домен), BEACH (Beige and Chediak-Higashi домен) и WD40-повторения (рис. 2) [5].

Рисунок 2 | Строение шести родственных белков семейства BEACH слизевика рода Dictyostelium [6].
На рис. 2 изображена диаграмма, наглядно показывающая общность строения шести белков семейства BEACH (в данном случае — шесть белков слизевика рода Dictyostelium). Зеленым цветом обозначен собственно BEACH-домен, наиболее консервативный во всех белках. «Овалами» у С-конца обозначены WD40-повторения — участки, предположительно вовлеченные в белок-белковые взаимодействия. Другими цветами показаны иные идентичные аминокислотные последовательности [6].
Сегодня известно, что белки данного семейства есть у разных представителей животного мира. Имея примерно одинаковое строение, они выполняют многочисленные и крайне сложные функции, обсуждение которых выходит за рамки данной темы.
Собственно LYST обеспечивает секрецию лизосомальных ферментов. Без белка-регулятора транспорт протеолитических ферментов невозможен, вследствие этого цитотоксическая активность фагоцитарных клеток нарушается. Этим также объясняется нарушение транспорта различных метаболитов (накопление в клетках гранул с меланином, что обеспечивает яркий клинический признак синдрома Чедиака-Хигаси), а также нарушения метаболизма некоторых других белков, что обусловливает клиническую картину заболевания и сопутствующие патологии, точный механизм развития которых все еще неизвестен [1,3].
Клиническая картина
Существует два основных клинических проявления синдрома Чедиака-Хигаси: «парциальный альбинизм» и рецидивирующие пиогенные инфекции. Оба, на первый взгляд, не связанных между собой критерия имеют единую причину — дефект LYST, вследствие чего нарушается транспорт меланина и лизосомальных ферментов.
Цвет волос пациента может быть от сероватого до буквально седого — зависит от этнической принадлежности пациента. Также нарушается пигментация радужной оболочки, следствием чего становится фоточувствительность и сниженная функция зрительного аппарата.
Рецидивирующие бактериальные (в том числе оппортунистические) инфекции часто поражают дыхательные пути, кожу. Возможны дефекты свертывающей системы из-за нарушенного синтеза тромбоцитов, проявляется это небольшими кровоподтеками (зачастую — на слизистых), однако иногда развивается полноценный геморрагический синдром.
Помимо этого возможны неврологические нарушения: атаксия, сенсорные расстройства, прогрессирующая нейродегенерация [1].
Первые проявления заболевания начинаются с раннего детства. Наиболее опасными осложнениями считаются инфекции, для предупреждения которых допустима профилактическая антибиотикотерапия. И все же большинство пациентов умирает в первые десять лет жизни. Наиболее распространенными причинами смерти являются кровотечения, инфекции или гемофагоцитарный лимфогистиоцитоз [7].
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз (ГФЛГ) — патологическое состояние, которое может быть вызвано разнообразными причинами, часто — первичными иммунодефицитами (к числу которых относится и синдром Чедиака-Хигаси). Суть данного состояния — гиперпродукция гистиоцитов и иммунокомпетентных клеток [5].
Как уже было сказано выше, мутация LYST нарушает цитотоксическую функцию клеток, но не метаболизм регуляторных факторов. В ответ на антиген развивается обыкновенная воспалительная реакция — однако элиминация чужеродного агента невозможна. В связи с этим продолжается продукция провоспалительных факторов (ИФН-γ, ФНО, интерлейкины), что увеличивает количество лимфоцитов, нейтрофилов, повышает активность макрофагов. Гиперпродукция цитокинов, не ингибированная по механизму обратной связи, иногда называется «цитокиновым штормом» [5].
Следствием этого является лимфогистиоцитарная инфильтрация различных тканей с развитием в них разнообразных повреждений, а макрофаги могут разрушать нормальные функционирующие клетки (в том числе — форменные элементы крови) [5].
Диагностика синдрома Чедиака-Хигаси и сопутствующих заболеваний
В диагностировании синдрома Чедиака-Хигаси особых сложностей не возникает. Как уже было сказано выше, основная задача клинициста — распознать врожденное иммунодефицитное состояние, дальнейшая диагностика основана на данных иммунограммы и генетического анализа. Достаточно специфичным признаком является накопление больших внутриклеточных везикул в различных клетках, в том числе — лейкоцитах [2].
Одним из наиболее серьезных осложнений при данном синдроме является развитие ГФЛГ. Диагностические критерии при этом патологическом состоянии можно представить следующим образом [5]:
- Наличие генетического дефекта, связанного с ГФЛГ (зачастую это первичные иммунодефициты).
- Наличие как минимум пяти из нижеперечисленных критериев:
- лихорадка;
- спленомегалия;
- цитопения хотя бы в двух клеточных популяциях:
- гипертриглицеридемия (> 3 ммоль/л) или гипофибриногенемия ( < 1,5 г/л);
- гиперферритинемия > 500 мкг/л;
- растворимые молекулы CD25 > 2400 Ед/мл;
- гемофагоцитоз в костном мозге, селезенке, лимфоузлах
- низкая (вплоть до полного отсутствия) цитотоксическая активность NK-клеток.
Важно отметить: ГФЛГ является настолько частым серьезным осложнением, что в современной литературе принято разделять синдром Чедиака-Хигаси на две формы: «классическую» (с развитием ГФЛГ) и «атипичную» (без такового) [5].
Лечение синдрома Чедиака-Хигаси и сопутствующих патологий
Как и в большинстве наследственных иммунодефицитов, вариантов терапии немного. Наиболее распространенным методом лечения врожденных иммунологических нарушений является пересадка гемопоэтических клеток; данный синдром не является исключением.
В 2007 году в «Bone Marrow Transplantation» появилась публикация, авторы которой сообщали о 35 случаях проведения пересадки гемопоэтических клеток пациентам с синдромом Чедиака-Хигаси [8]. Перед проведением операции лечение осуществлялось в лучшем случае патогенетическое, призванное замедлить прогрессирование заболевания, избежать осложнений и подготовить пациента к пересадке гемопоэтических клеток.
Всего 13 пациентов получили материал от HLA-идентичного донора (родного брата или сестры), 10 — от родственника, еще 12 — от несвязанного донора. По результатам проведенной терапии, 27 (77 %) из 35 пациентов достигли ремиссии, однако 5-летняя выживаемость составила 62 % (22 пациента).
Наиболее распространенными причинами смерти были посттрансплантационные осложнения и хронические заболевания. Среди умерших — большинство получило аллотрансплантат от не полностью подходящего донора.
Таким образом, исследователи пришли к выводу, что подобная методика может быть достаточно эффективной, если донор и реципиент являются родственниками. Также следует отметить, что шесть пациентов на момент проведения операции были старше девяти лет, что свидетельствует о существенных успехах в сдерживании и предотвращении развития осложнений у пациентов (одному из реципиентов на момент трансплантации было 19 лет).
На современном этапе пересадка гемопоэтических клеток является наиболее эффективным способом лечения синдрома Чедиака-Хигаси [9]. Вероятно, в будущем станет возможна коррекция непосредственно LYST с помощью генно-инженерных методик (например, подобная терапия уже существует для хронической гранулематозной болезни — другого наследственного иммунодефицитного состояния [10]), однако пока что такой альтернативы для синдрома Чедиака-Хигаси нет.
Часто синдром Чедиака-Хигаси осложняется ГФЛГ — тяжелым состоянием, которое требует немедленного лечения. Среди наиболее эффективных методов терапии в гайдлайне 2004 года описаны химиотерапия и пересадка гемопоэтических клеток [11].
Также на протяжении лечения, до и после него в профилактических целях используется антибиотикотерапия [1,11].
Читайте также:
- Ультрафильтрация с инотропной поддержкой. Пример комбинации добутамина и гемофильтрации
- Диагностика синдрома повторного сотрясения головного мозга по КТ, МРТ
- Обсуждение применения трансплантатов при лечении вращательной манжеты плечевого сустава
- Методы лечения артроза тазобедренного сустава. Показания к пункции и введению лекарств в сустав
- Синдром Страхана-Скотта (Strachan-Scott)