Апластическая анемия (миелоидная аплазия) - история изучения, классификация
Добавил пользователь Alex Обновлено: 28.01.2026
Миелодиспластический синдром (МДС) - это не одна какая-то болезнь, это целая группа различных патологических состояний костного мозга (КМ), отнесенных к гематологии, но пока не причисленных к лейкозам, хотя болезнь оставляет высокий риск перехода в более тяжелую форму (лейкоз).
Суть МДС заключается в нарушении костномозгового кроветворения на миелоидной линии в отношении какого-то одного клона клеток или затрагивающего несколько популяций. В любом случае для миелодиспластического синдрома характерным признаком будет изменение качественного и количественного состава периферической крови.
Коротко о гемопоэзе
Кроветворение (гемопоэз) - процесс, проходящий много стадий, на каждой из которых клетки крови приобретают новые качества (дифференцируются). Конечным результатом этого процесса является выход в периферическую кровь зрелых (или созревающих, но уже имеющих определенные «навыки»), полноценных, способных осуществлять свои функциональные задачи, форменных элементов крови:
- Красных кровяных телец - эритроцитов;
- Белых клеток - лейкоцитов;
- Кровяных пластинок (бляшек Биццоцеро) - тромбоцитов.
Кроветворение начинается от стволовой клетки, способной, дифференцироваться и давать жизнь всем линиям (росткам) гемопоэза. Миелоидный и лимфоидный ростки пошли от специализированных, обладающих высокой пролиферативной активностью, способных к дифференцировке плюрипотентных клеток.
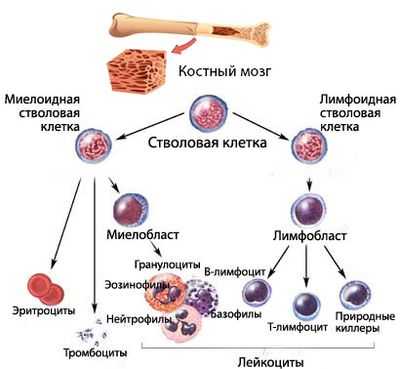
Сбой кроветворения в миелоидном направлении приводит к тому, что сам аномальный клон в некоторой степени теряет возможность продолжать линию (воспроизводить потомство, поэтому количество клеток того ростка, на котором возникла проблема, падает). Естественно, нарушается и созревание полноценных клеток. В результате этого, уменьшается численность одной или нескольких популяций форменных элементов, а также, ввиду ухудшения качества клеток, не в лучшую сторону меняются их функциональные возможности.
Вытекающие из подобных событий последствия - синдром, имеющий различные варианты клинических проявлений, то есть, представляющий собой группу гетерогенных патологических состояний, которая и названа миелодиспластическим синдромом.
Позиция МДС в Международной классификации болезней
Международная классификация болезней десятого пересмотра (МКБ-10), принятая Всемирной организацией здравоохранения (ВОЗ) в Швейцарии Женева, 1989), вступила в силу на территории Российской Федерации в 1997 году. Между тем, в отношении многих патологических состояний в 2010 году были внесены изменения. Нововведения коснулись и гематологической патологии, в том числе, миелодиспластического синдрома. По МКБ-10 в блок диагнозов D37-D48 МДС входит под своим кодом - D46, который имеет 7 или 9 вариантов определений заболеваний или диагнозов (в России, наряду с классификацией ВОЗ, могут использоваться и другие классификации, например, FAB, где вообще только 5 вариантов, поэтому в разных справочниках кодирование также может иметь отличия):
пример МДС с избытком бластов
Примечание: так часто встречающееся определение «рефрактерная» в данном случае объясняет безуспешность лечения железосодержащими и витаминными лекарственными средствами. Рефрактерная анемия устойчива к подобным мерам воздействия, не реагирует на них и нуждается в других терапевтических мероприятиях.
Общая характеристика синдрома
Аномалия генетического материала на уровне полипотентной кроветворной стволовой клетки, мутация ее, а также клеток предшественниц кроветворения, наличие генетически неполноценных клонов приводят к тому, что в клеточном звене системы иммунитета происходят существенные изменения, глубина которых, однако, зависит от того, по каким линиям (одной или нескольким?) пошли нарушения в кроветворении. В зависимости от этого можно ожидать в крови:
- Моноцитопению (уменьшение клеток одного вида);
- Бицитопению (нарушения идут в двух ростках);
- Панцитопению (сбой пошел в трех направлениях, поэтому резко снижено количество белых и красных клеток крови, а также тромбоцитов).
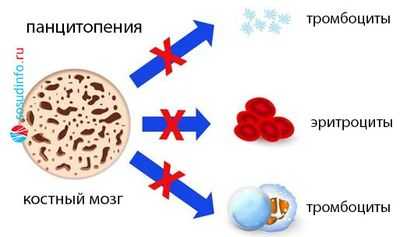
В КМ - аналогично: нормоклеточность, гиперклеточность либо гипоклеточность (миелограмма покажет, какой росток пострадал).
Клинические проявления описываемого синдрома также соответствуют причине, спрятанной на уровне кроветворения:
- Анемия;
- Геморрагический синдром (при снижении численности и нарушении функции тромбоцитов);
- Сочетание анемического и геморрагического синдромов;
- Инфекционный синдром (реже);
- Увеличение селезенки, лимфаденопатия, постоянное повышение температуры тела (эти симптомы присутствуют не так и часто, поэтому относятся к факультативным признакам).
Между тем, опираясь на данные многочисленный исследований МДС (изменение численности и морфологических характеристик клеток крови и костного мозга), гематологи пришли к выводу, что рано или поздно конечным итогом миелодиспластического синдрома станет острый или хронический миелоидный лейкоз (ОМЛ либо ХМЛ), а все эти анемии (рефрактерные) являются лишь промежуточным (временным) состоянием болезни. В связи с этим МДС нередко называют «предлейкозом», «предлейкемией», «тлеющим» или «дремлющим» лейкозом. Все зависит от количества миелобластов - клеток-родоначальниц гранулоцитарного ряда.
Если рефрактерная анемия протекает с избытком бластов (>20% по данным ВОЗ или >30% согласно классификации FAB), то гематологи склоняются к диагнозу - миелоидный лейкоз. В ситуациях, когда численность бластных клеток вплотную не подходит к этому порогу, диагноз пациента остается прежним - миелодиспластический синдром.
Патологическое состояние главного кроветворного органа может сформироваться у человека в любом возрастном периоде (от грудного - до глубокой старости). У детей болезнь чаще всего дебютирует между 3 и 5 годами, хотя, в целом, в детском возрасте риск заболеть совсем низкий. Среди взрослых самыми уязвимыми становятся пожилые люди (60 лет и старше). Например, такой распространенной и рискующей перейти в острый лейкоз форме, как РЦМД, наиболее подвержены люди в возрасте от 70 до 80 лет. Общая частота встречаемости миелодиспластического синдрома колеблется в пределах 3-5 случаев на 100 тысяч населения (не так и редко), причем, мужчины страдают данной патологией несколько чаще, нежели женщины.
Причина первичных форм заболевания остается невыясненной. Основными вероятными «виновниками» вторичного МДС считаются:
- Воздействие ионизирующего излучения;
- Влияние антропогенных неблагоприятных факторов окружающей среды (химических соединений, созданных человеком);
- Последствия химио- и радиотерапии (после лечения опухолевых процессов);
- Инфекционные агенты (бактерии, вирусы).
Следует отметить, что до сих пор МДС, передаваемого по наследству или возникающего в кругу близких родственников, отмечено не было, однако из наблюдений определена группа пациентов, имеющих повышенный риск формирования синдрома. Это дети и взрослые люди, страдающие болезнью Дауна, анемией Фанкони, синдромами Луи-Бар и Блума.
Лечатся все по-разному
Следует сразу настроить пациента, что лечение МДС не будет одинаковым для всех его разновидностей. Набор терапевтических мероприятий рассматривается в индивидуальном порядке, исходя из формы болезни и категории риска, которой принадлежит пациент (согласно клинической классификации Международной Прогностической Системы - IPSS для МДС: низкий, промежуточный 1 и 2, высокий). Словом, существуют определенные каноны, которых доктор придерживается, прежде чем приступить к непосредственному лечению. К примеру:
- Люди, не перешагнувшие 60-летний рубеж, имеющие минимальные признаки болезни, но отнесенные к категории промежуточного или высокого риска с ожидаемой выживаемостью 0,3 - 1,8 года, подвергаются высокоинтенсивной терапии;
- Пациенты, принадлежащие к группе промежуточного и низкого риска с ожидаемой выживаемостью 5-12 лет, проходят лечение низкой интенсивности;
- Молодые люди и больные среднего возраста (до 60 лет) с относительно неплохими показателями (ожидаемая выживаемость от полугода до 5 лет) первоначально лечатся по схемам низкой интенсивности, хотя в любой момент им грозит оказаться в группе, получающей более жесткое лечение (высокие дозы химиотерапии, пересадка КМ).
Таким образом, схемы лечения миелодиспластического синдрома довольно сложны и знает их только врач, получивший в свое время определенную специализацию (гематолог). Он в своей лечебной тактике опирается на рекомендации, разработанные Британским комитетом по стандартизации в гематологии (редакция 2009 года). Читателю же, на наш взгляд, достаточно познакомиться с основными методами проведения терапевтических мероприятий, особо не вникая в тонкости, не ставя диагноз и не причисляя себя или своих близких к той или иной группе риска. И еще, наверное, не помешает знать, что:
- Лечение высокой интенсивности - это, во-первых, обязательное пребывание в специализированном стационаре, во-вторых, назначение высоких доз химиотерапии и, возможно, подготовка к пересадке стволовых клеток и сама пересадка;
- Низкоинтенсивная терапия подразумевает пребывание в больнице (или даже в условиях дневного стационара) время от времени для получения заместительной терапии, низких доз химиопрепаратов, симптоматического лечения.
К сожалению, способа избавиться от такого тяжелого недуга, как МДС, раз и навсегда, пока не придумали. Разве что пересадка главного кроветворного органа (костного мозга) могла бы решить проблему, однако она тоже сопряжена с определенными трудностями (иммунологическое типирование, поиск совместимого донора, высокая стоимость операции, если искать донора по всему миру). Правда, в последние годы, как на территории Российской Федерации и ближайшей соседки - Беларуси, так и на территории других государств бывшего СССР, создаются новые лаборатории тканевого типирования, объединяющие свои реестры в единый банк, чтобы иметь возможность помочь друг другу. На них и возлагаются будущие надежды.
Лечение

Если врач считает, что патологический процесс идет как бы доброкачественно (если можно так выразиться), с небольшим количеством бластов, то больные группы низкого риска, периодически получающие заместительное и поддерживающее лечение (эритроцитарную массу, тромбовзвесь), могут довольно продолжительное время работать и вести почти привычный образ жизни. В основном, лечение таких больных выглядит следующим образом:
- Больной направляется в стационар, чтобы не допустить значительного снижения гемоглобина и развития тяжелого анемического синдрома, поэтому борьбу с ним (анемическим синдромом) считают первостепенной задачей (переливание эритроцитарной массы, заготовленной от доноров);
- Не упускается из виду и такое проявление МДС, как геморрагический синдром, возникающий на почве снижения числа и функциональной неполноценности тромбоцитов. В принципе, симптоматическая терапия, которая позволяет удерживать количество форменных элементов на нужном уровне (гемотрансфузии - эрмасса, тромбовзвесь и т. д.), в общем-то, всегда присутствует в схеме лечения больных, имеющих относительно благоприятную форму болезни;
- Получая от случая к случаю донорские эритроциты, организм больного начинает перегружаться железом, что ликвидируется применением медикаментозных средств, образующих комплексы с этим химическим элементом (эксиджад, десферол);
- Иной раз больные нуждаются в назначении низких доз «химии» (цитарабин, децитабин), а также иммунодепрессивных средства для предотвращения иммунной агрессии против костного мозга (леналидомид), с добавлением к ним ATG (антимоноцитарный глобулин) и циклоспорина;
- Присоединение инфекционного агента требует лечения антибиотиками и противогрибковыми препаратами.
Гораздо сложнее лечить формы миелодиспластического синдрома с избытком бластов, входящие в категорию высокого риска, когда химиотерапевтические препараты почти не приносят желаемого результата и не «отправляют» больного в долгосрочную ремиссию. Однако это не значит, что от них отказываются вовсе, ведь новые, недавно разработанные лекарства, дают некоторую надежду в отношении МДС и даже применяются для лечения ОМЛ (острого миелобластного лейкоза). Однако при таких обстоятельствах существуют рекомендации разработчиков - применять подобные средства для лечения больных, не достигших 60-летнего возраста и имеющих неплохой иммунологический статус, в противном случае - есть риск развития серьезных осложнений, способных преждевременно прервать жизнь.
Пересадка стволовых клеток (возможна тоже только до 60 лет) на сегодняшний день - единственный способ избавить человека от страданий на долгие-долгие годы. К сожалению, трансплантация КМ - операция хоть и несложная в техническом плане, но трудновыполнимая в плане подбора по лейкоцитарной системе HLA совместимого с реципиентом (больным) донора (идентичными, то есть, имеющими абсолютно одинаковый набор генов являются только однояйцевые близнецы - это идеальные доноры друг другу).
Частные симптомы и диагностика
Клинические проявления и степень их выраженности по причине многообразия форм МДС позволяют себе широкие вариации. Случайной находкой синдром выступает редко (это бывает, если человек неплохо себя чувствует, а анализы назначаются в силу других обстоятельств). В основном же, больные направляются в поликлинику с определенными жалобами (постоянное ощущение усталости, одышка, физическая слабость, головокружения, частые подъемы температуры тела), где после тестирования крови становятся очевидными и другие признаки миелодиспластического синдрома:
- Цитопения (снижение количества полноценных форменных элементов крови);
- Анемия (низкий гемоглобин, мало эритроцитов), которая и определяет симптомы, заставившие пойти к врачу;
- Нейтропения (недостаточное содержание в крови нейтрофильных лейкоцитов, обладающих способностью поглощать бактериальные клетки в очаге воспаления - она становится причиной частых инфекций и лихорадки);
- Тромбоцитопения (уменьшение численности тромбоцитов, что обуславливает появление геморрагического синдрома - кровотечений, мелкоточечных подкожных кровоизлияний, синяков).
Между тем, отдельные пациенты относительно долго могут жить и не подозревать, что здоровье «пошатнулось». И тогда МДС становится случайной находкой уже на стадии проведения общего анализа крови.
Чаще всего поводом все же обратиться в поликлинику служат жалобы больного, которые в наибольшей степени связаны с анемией. Пробовать повысить уровень красного пигмента крови (Hb) и содержание красных кровяных телец (Er) препаратами железа и витаминами бесполезно, лечение успехов не приносит, ведь анемия при МДС - рефрактерная. При подозрении на МДС, которое возникает в ходе проведения общего анализа крови (ОАК), добавляются другие исследования:
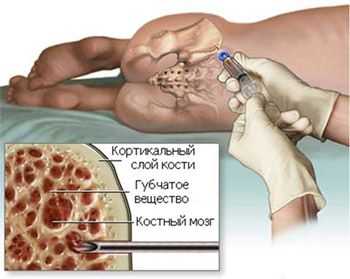
- Подсчет молодых форм красного ростка, которым уже «позволено» присутствовать в циркулирующей крови - ретикулоцитов, они «подскажут», насколько быстро идет процесс воспроизводства новых зрелых кровяных телец;
- Цитологическое исследование аспирата КМ (у пожилых пациентов данный тест не принадлежит к обязательным анализам);
трепанбиопсия костного мозга
Трепанобиопсия (процедура обязательна для всех больных) - после изучения морфологических особенностей гистологический анализ развеет сомнения или подтвердит подозрения;
Безусловно, диагностика миелодиспластического синдрома, начинается с жалоб больного и ОАК, но в дальнейшем опирается на более сложные лабораторные исследования. Здесь врачу есть над чем подумать, чтобы правильно оценить нарушения кроветворения, ведь изменения клеточного состава и морфологических особенностей клеток крови и костного мозга могут быть весьма многочисленны и многообразны. Впрочем, как и сама болезнь…

костный мозг при МДС
Прогноз и рекомендации
Прогноз в отношении продолжительности жизни при миелодиспластическом синдроме не очень оптимистичный, хотя многое зависит от разновидности болезни, степени риска и возрастной категории больного. В целом, пациенты, строго выполняющие рекомендации лечащего врача и получающие периодически поддерживающее лечение, могут рассчитывать прожить пять, а то и десять лет. Однако активное течение злокачественной формы болезни оставляет мало шансов - если не был найден донор и не пересажена стволовая клетка, жизнь может прерваться на 1-2 году от начала патологического процесса. Причиной смерти в большинстве случаев становится острый миелоидный лейкоз, который развился на почве вторичного МДС.
В заключение хочется дать совет людям, столкнувшимся с подобной проблемой и желающим продлить свою жизнь или жизнь близким: никогда не слушать рекомендации того, кто почерпнул сведения о болезнях из сомнительных источников (подобная информация вовсю «гуляет» на просторах Интернета) и возомнил себя доктором. Ни народными средствами, ни специальными физическими упражнениями миелодиспластический синдром не лечится. Нужно следовать рекомендациям врача и тогда, возможно, лечение будет успешным.
Анемия Даймонда-Блекфена ( Наследственная парциальная красноклеточная аплазия )
Анемия Даймонда-Блекфена - это наследственная форма красноклеточной аплазии с достоверно неизученным типом наследования (предполагается аутосомно-доминантный тип наследования, встречающийся у четверти больных). Симптомами заболевания являются анемические проявления, возникающие, как правило, на протяжении первого года жизни - бледность, слабость, повышенная утомляемость, уменьшение в крови количества эритроцитов. Диагностика производится на основании данных общего анализа крови, исследования уровня эритропоэтинов, биопсии и микроскопии костного мозга, в четверти случаев информативно генетическое исследование. Лечение осуществляется с помощью гемотрансфузий, глюкокортикостероидов.
МКБ-10


Общие сведения
Анемия Даймонда-Блекфена (наследственная парциальная красноклеточная аплазия) представляет собой генетическое поражение системы крови, при котором нарушается образование эритроцитов. Название патологии было дано по фамилиям врачей, которые в 1938 году совместно обследовали четверых детей с симптомами сильной анемии наследственного характера.
Это состояние относится к очень редким, на сегодняшний день достоверно описано около 500 случаев. Подсчитано, что встречаемость анемии Даймонда-Блекфена составляет около 4-6:1000000, ей в одинаковой степени подвержены как мальчики, так и девочки. Несоответствие количества доказанных случаев с рассчитанной встречаемостью объясняется тем, что у определенной части больных ошибочно диагностируется либо эритромиелобластный лейкоз, либо приобретенные формы парциальной красноклеточной аплазии. Наиболее распространенная форма заболевания наследуется по аутосомно-доминантному принципу, однако это объясняет только 25% всех случаев, тогда как по поводу остальных вариантов пока данных нет.

Причины
Непосредственной причиной четверти случаев анемии Даймонда-Блекфена служит мутация в гене RPS19 расположенном в 19-й хромосоме, который кодирует важный рибосомальный белок S19. Последний входит в состав малой (40S) субъединицы рибосомы человека. Такая мутация наследуется по аутосомно-доминантному механизму со встречаемостью 6 случаев на миллион человек. В других случаях были обнаружены мутации иных генов, однако они так или иначе связаны с рибосомальными белками - это гены RPS7, RPS24, RPL5, RPL32A и ряд других.
Распространенность таких мутаций, характер их наследования, доля в общем количестве заболевших анемией Даймонда-Блекфена, их влияние на прогноз и исход патологии в настоящий момент остается объектом изучения врачей-генетиков. Также интерес представляет собой вопрос, почему мутации генов рибосомальных белков оказывают влияние именно на эритропоэз и почти не влияют на другие ростки кроветворения.
Патогенез
Существует несколько теорий, пытающихся объяснить торможение образования эритроцитов в красном костном мозге. Наиболее распространенные указывают в качестве возможных причин анемии Даймонда-Блекфена дефекты микроокружения клеток-предшественников эритроцитов, их внутренние аномалии, супрессию со стороны иммунной системы или гуморальные факторы, останавливающие созревание эритробластов. Ни одна из теорий на сегодняшний день не получила достоверного и однозначного подтверждения.
При этом заболевании в красном костном мозге наблюдается неуклонное снижение эритроидных единиц, причем примерно в трети случаев данный процесс начинается во время внутриутробного развития, что позволяет диагностировать анемию Даймонда-Блекфена сразу после рождения. Соответственно, начинает снижаться количество выделяемых в кровь эритроцитов, в костном мозге накапливаются эритробласты, что может вводить в заблуждение (подобные изменения характерны для лейкоза). При этом у младенцев уровень фетального гемоглобина может не снижаться, поэтому данный показатель не считается диагностическим в случае анемии Даймонда-Блекфена. Возникает компенсаторный рост уровня эритропоэтинов в крови, однако они в данном случае не способны увеличить скорость образования эритроцитов. В конечном итоге развивается выраженная анемия.
Симптомы анемии
На первый план при синдроме Даймонда-Блекфена выступают анемические симптомы - бледность, слабость ребенка, у грудных младенцев часто развивается гипотрофия, наблюдается недобор массы. Примерно у половины больных помимо нарушений со стороны крови также возникает ряд физических отклонений - микроцефалия, гипертелоризм, птоз век, микрогнатия. Возможны аномалии скелета - увеличение размера лопаток и кистей, отсутствие некоторых пальцев, задержка роста костной ткани. В некоторых случаях возможны такие нарушения как «заячья губа».
Поражаются и органы зрения - развивается косоглазие, глаукома, катаракта. Многие указанные симптомы возникают в раннем возрасте ребенка и усугубляются выраженной анемией, поэтому своевременно начатое лечение может значительно ослабить или даже устранить многие из них.
В отличие от транзиторных и приобретенных анемий, синдром Даймонда-Блекфера незначительно влияет на работу печени и селезенки - их заметное увеличение может возникать лишь на конечных стадиях заболевания или в результате осложнений гемотрансфузионной терапии.
Диагностика
При осмотре ребенка, больного анемией Даймонда-Блекфена, определяется бледность кожных покровов, синюшность слизистых, на голове заметны венозные сосуды. Также могут наблюдаться сопутствующие заболеванию физические отклонения (микроцефалия, гипертелоризм и другие), при взвешивании часто выявляется недобор массы тела. Методы лабораторной диагностики включают:
- Анализы крови. ОАК показывает картину нормохромной анемии, часто макроцитарного характера, резкое снижение количества ретикулоцитов. В некоторых случаях наблюдается гранулоцитопения и тромбоцитопения, однако это не может служить достоверным диагностическим критерием анемии Даймонда-Блекфена. При биохимическом исследовании крови выявляется резкое увеличение уровня эритропоэтина.
- Биопсия костного мозга. При микроскопическом исследовании биоптата костного мозга выявляется в целом нормоклеточный тип с выраженным снижением клеток эритроидного ряда. При этом при некоторых формах анемии Даймонда-Блекфена в костном мозге могут накапливаться эритробласты, что часто ведет к ошибочной диагностике острого миелолейкоза.
- Генодиагностика. Современная генетика методом прямого секвенирования последовательности определяет мутации только одного гена, ассоциированного с анемией Даймонда-Блекфена - RPS19. С помощью данного метода диагностики мутации гена обнаруживаются только в 25-30% случаев клинически выявленного заболевания.
Лечение анемии Даймонда-Блекфена
При выраженной анемии показаны гемотрансфузии или переливание эритроцитарной массы для восполнения угрожающего жизни дефицита эритроцитов. В некоторых случаях переливание крови может потребоваться неоднократно - например, при отсутствии эффекта от глюкокортикостероидной терапии. В таких ситуациях необходимо принимать меры в отношении профилактики поражения печени и селезенки избытком железа и других посттрансфузионных осложнений - например, назначать хелационную терапию дефероксамином.
Основными препаратами для лечения анемии Даймонда-Блекфена являются глюкокортикостероиды (преднизолон, метилпреднизолон и другие). Терапию начинают с повышенных (ударных) дозировок препаратов внутрь, затем постепенно снижая их до уровня поддерживающей дозы - при этом одновременно должен расти уровень гемоглобина и улучшаться картина крови (в кровотоке появляются ретикулоциты, снижается количество макроцитов).
В зависимости от динамики заболевания терапию глюкокортикостероидами производят при помощи двух основных схем - в режиме пульс-терапии (до 7-ми дней приема повышенных доз с последующим двух-трех недельным перерывом) и поддерживающей терапии с ежедневным приемом небольших количеств глюкокортикостероидов. Выбор той или иной схемы зависит от реакции организма больного на препараты, наличия и выраженности побочных явлений, эффективности лечения. Описаны несколько случаев лечения анемии Даймонда-Блекфена с положительным исходом посредством пересадки костного мозга от близкого родственника.
Прогноз
Прогноз анемии Даймонда-Блекфена во многом неопределенный по причине слабого понимания процессов, которые приводят к ее развитию. Обширное исследование, в ходе которого изучили историю жизни более чем 200 больных, выявило, что почти у четверти детей, которых лечили посредством гемотрансфузий и назначения глюкокортикостероидов, еще до подросткового возраста возникла спонтанная ремиссия с восстановлением адекватного уровня гемоглобина. При этом больше половины детей остались зависимыми как от переливаний крови, так и от использования метилпреднизолона и после подросткового возраста. Оставшиеся 25% больных умерли еще в детстве, несмотря на все предпринятые терапевтические меры.
Во многом на прогноз влияет наличие или отсутствие сопутствующих физических отклонений и степень их выраженности. Лечение ростовыми факторами, препаратами железа и другими традиционными при анемии средствами неэффективно и может еще больше усилить нагрузку на печень и селезенку.
2. едеральные клинические рекомендации по диагностике и лечению анемии Даймонда-Блекфена у детей. - 2015.
3. Анемия Даймонда-Блекфана: модель трансляционного подхода к пониманию заболеваний у людей/ Влахос А.Бланк Л.Липтон Дж.М.// Российский журнал детской гематологии и онкологии. - 2014.
Миелодиспластический синдром
Миелодиспластический синдром - группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение - переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.

Миелодиспластический синдром - группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.

Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома - курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие - при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким - около 8 месяцев. Вероятность отторжения костного мозга после трансплантации - около 10%.
Апластическая анемия ( Гипопластическая анемия )
Апластическая анемия - угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.


Апластическая (гипопластическая) анемия - тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10-25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму - постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией - снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Классификация
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии - низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов - стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия - при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Осложнения
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений - кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз и профилактика
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75-90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.
Апластическая анемия у взрослых
Приобретённая (идиопатическая) апластическая анемия - панцитопения с гипоцеллюлярным костным мозгом в отсутствие патологической инфильтрации и повышения количества ретикулиновых волокон.
Апластическая анемия - гетерогенное заболевание. В большинстве случаев (70-80%) этиология остается неясной - идиопатическая апластическая анемия. Примерно в 15-20% наблюдаются конституциональные/врожденные варианты апластической анемии. В некоторых случаях удается установить причину апластической анемии - лекарственное воздействие (таблица 1), инфекцию и др.
Таблица 1. Препараты, использование которых может быть ассоциировано с АА.
| Группа | Препараты |
| Антибиотики | Хлорамфеникол, Сульфаниламиды, Ко-тримоксазол, Линезолид |
| Противовоспалительные препараты | Препараты золота, Пеницилламин, Индометацин, Диклофенак, Напроксен, Пироксикам, Сульфасалазин |
| Противосудоржные препараты | Карбамазепин, Фенитоин |
| Тиростатики | Тиоурацил, Карбимазол |
| Антидепрессанты | Фенотиазины |
| Гипогликемические средства | Хлорпропамид, Толбутамид |
| Противомалярийные препараты | Хлорохин |
| Другие | Аллопуринол, Тиазиды |
Код(ы) МКБ-10:
| МКБ-10 | |
| Код | Название |
| D 61.3 | Идиопатическая апластическая анемия |
Дата разработки/пересмотра протокола: 2015 год (пересмотр 2018 г.)
Сокращения, используемые в протоколе:
полимеразная цепная реакция
Пользователи протокола: врачи общей практики, терапевты, гематологи, гинекологи.
Категория пациентов: взрослые, беременные.
Шкала уровня доказательности [1]:
Читайте также: