Болезнь Эбштейна. Анатомия и формы болезни Эбштейна
Добавил пользователь Skiper Обновлено: 01.02.2026
МКБ-10
Общие сведения
Аномалия Эбштейна - врожденная патология трехстворчатого предсердно-желудочкового клапана, сопровождающаяся аномальным положением створок, приводящим к образованию над ними атриализованной части правого желудочка, составляющей одно целое с правым предсердием. Впервые в кардиологии аномалия была описана немецким врачом W. Ebstein в 1866 г. Частота аномалии Эбштейна среди различных врожденных пороков сердца составляет 0,5-1%. Аномалии Эбштейна часто сопутствуют другие пороки сердца - дефект межпредсердной перегородки (ДМПП), открытый артериальный проток (ОАП), дефект межжелудочковой перегородки (ДМЖП), стеноз или атрезия легочной артерии, митральный стеноз или недостаточность, синдром WPW.
Причины
Формирование аномалии Эбштейна связывается с поступлением лития в организм плода на ранних этапах эмбриогенеза. Также к возникновению данного и других пороков сердца могут приводить инфекционные заболевания беременной (скарлатина, корь, краснуха), тяжелые соматические заболевания (анемия, сахарный диабет, тиреотоксикоз), употребление алкоголя и лекарственных средств, обладающих тератогенным воздействием, патология гестации (токсикозы, угроза самопроизвольного выкидыша и пр.).
Важную роль в развитии аномалии Эбштейна играет отягощенная наследственность по ВПС - наряду со спорадическими, встречаются и семейные случаи аномалии.
Классификация
Выделяют четыре анатомических варианта (типа) аномалии Эбштейна:
- I - передняя створка трикуспидального клапана большая и подвижная; септальная и задняя створки отсутствуют или смещены;
- II - присутствуют все три створки предсердно-желудочкового клапана, однако они относительно малы и смещены по направлению к верхушке сердца.
- III - хорды передней створки трикуспидального клапана укорочены и ограничивают ее движение; септальная и задняя створки недоразвиты и смещены.
- IV - передняя створка трикуспидального клапана деформирована и смещена в сторону выходного тракта правого желудочка; септальная створка образована фиброзной тканью, задняя - недоразвита или полностью отсутствует.
Особенности гемодинамики при аномалии Эбштейна
Анатомическую основу аномалии Эбштейна составляет неправильное расположение трехстворчатого клапана, при котором его створки (обычно задняя и перегородочная) оказываются деформированы и смещены в полость правого желудочка. В этом случае створки клапана крепятся ниже фиброзного кольца, иногда на уровне выходного отдела правого желудочка. Смещение клапана сопровождается атриализацией правого желудочка, т. е. состоянием, при котором часть правого желудочка составляет продолжение и единую полостью с правым предсердием.
Таким образом, смещение створок обусловливает деление правого желудочка на 2 функциональные части: надклапанную атриализованную, образующую общую полость с правым предсердием, и подклапанную - меньшую по размерам, функционирующую как правый желудочек. При этом правое предсердие и атриализованная часть правого желудочка значительно расширены, а полость правого желудочка - уменьшена.
Гемодинамические нарушения, сопутствующие аномалии Эбштейна, зависит от степени трикуспидальной недостаточности, размеров функционирующего правого желудочка и величины сброса крови справа налево через межпредсердные коммуникации.
Электрические процессы в правом предсердии, состоящем из двух частей, не синхронизированы: сокращение собственно правого предсердия происходит в систолу предсердий, а атриализованной части правого желудочка - в систолу желудочков. Вследствие трикуспидальной недостаточности происходит регургитация венозной крови обратно в правое предсердие; ударный объем правого желудочка снижается, что сопровождается уменьшением легочного кровотока. Правое предсердие дилатируется и гипертрофируется, давление в нем прогрессирующе повышается, вызывая возникновение венозно-артериального шунта через дефект в межпредсердной перегородке.
Право-левый сброс крови играет двоякую роль: с одной стороны он позволяет избежать перегрузки правого предсердия и компенсирует порок; с другой стороны - способствует развитию артериальной гипоксемии.
Симптомы аномалии Эбштейна
В зависимости от тяжести нарушений гемодинамики выделяют 3 стадии течения аномалии Эбштейна: I — бессимптомную (встречается редко); II — стадию выраженных гемодинамических расстройств (IIa — без нарушений сердечного ритма; IIб — с нарушениями сердечного ритма), III — стадию стойкой декомпенсации.
Самые тяжелые формы аномалии Эбштейна могут вызывать внутриутробную гибель плода. При благоприятном варианте порока его течение долго время остается бессимптомным; физическое развитие детей соответствует возрасту. В типичных случаях аномалия Эбштейна проявляется в раннем детстве, иногда в первые месяцы жизни ребенка.
Клинические симптомы включают диффузный цианоз, плохую переносимость физической нагрузки, боли в сердце, приступы сердцебиения. У 25-50% пациентов с аномалией Эбштейна отмечается пароксизмальная наджелудочковая тахикардия, у 14% из них - синдром WPW. При внешнем осмотре обращает внимание изменения концевых фаланг пальцев по типу «барабанных палочек» и ногтей в виде «часовых стекол», «сердечный горб».
При аномалии Эбштейна может рано развиваться правожелудочковая недостаточность - одышка, увеличение печени, набухание и пульсация шейных вен. Часто отмечается артериальная гипотония. Течение аномалии Эбштейна неуклонно прогрессирующее. Причиной смерти больных старшего возраста чаще всего служат сердечная недостаточность и тяжелые нарушения ритма.
Диагностика аномалии Эбштейна
Пациентам с подозрением на аномалию Эбштейна проводится консультация кардиолога и кардиохирурга, ЭКГ, рентгенография грудной клетки, ЭхоКГ, фонокардиография. Перкуторно определяется увеличение размеров сердца вправо, при аускультации выслушивается характерный трех- или четырехтактный ритм, систолический и диастолический шум справа от мечевидного отростка, расщепление II тона.
ЭКГ-данные включают отклонение ЭОС вправо, признаки гипертрофии и дилатации правого предсердия, пароксизмальную желудочковую экстрасистолию и предсердную тахикардию (синдром WPW), трепетание предсердий, мерцательную аритмию, полную (неполную) блокаду правой ножки пучка Гиса. Фонокардиограмма при аномалии Эбштейна характеризуется наличием систолического шума в проекции правого желудочка; запаздыванием I тона; раздвоенным II тоном; III, IV тонами большой амплитуды.
Рентгенологические признаки, свидетельствующие в пользу аномалии Эбштейна, представлены резким увеличением правых отделов сердца, шаровидной формой тени сердца, повышенной прозрачностью легочных полей.
При проведении эхокардиографии видно смещение книзу створок трикуспидального клапана, увеличение размеров правого предсердия, замедленное смыкание трехстворчатого клапана, смещение створок, наличие атриализированного правого желудочка, шунтирующий проток крови справа налево через ДМПП (по данным допплер-эхокардиографии). Фетальная ЭхоКГ, выполненная в пренатальном периоде, позволяет диагностировать аномалию Эбштейна в 60% случаев.
Для уточнения формы и степени тяжести аномалии Эбштейна проводится МРТ, зондирование полостей сердца, вентрикулография. Диагноз аномалии Эбштейна требует дифференциации с экссудативным перикардитом, миокардитом Абрамова-Фидлера, изолированным ДМПП и пульмональным стенозом, тетрадой Фалло.
Лечение аномалии Эбштейна
Медикаментозная терапия при аномалии Эбштейна проводится с целью лечения сердечной недостаточности и устранения аритмий. Показаниями к хирургической коррекции аномалии Эбштейна служат наличие жалоб, недостаточности кровообращения и нарушений ритма сердца. Оптимальным для операции является возраст 15-17 лет, при тяжелой форме порока вмешательство проводится в более ранние сроки.
Радикальная корригирующая операция при аномалии Эбштейна включает пластику или протезирование трикуспидального клапана, пластику ДМПП, ликвидацию атриализованного правого желудочка. В некоторых случаях целесообразным является выполнение операции Фонтена. Иногда на первом этапе для увеличения легочного кровотока и уменьшения гипоксемии прибегают к наложению анастомоза по Блэлоку-Тауссигу, наложению двунаправленного кава-пульмонального анастомоза.
Добавочные пути проведения импульса при WPW синдроме подвергаются радиочастотной абляции. Для лечения аритмий применяется имплантация кардиостимуляторов или кардиовертер-дефибрилляторов.
Прогноз
Естественное течение аномалии Эбштейна зависит от морфологического субстрата порока. На первом году жизни от тяжелейшей сердечной недостаточности или фибрилляции желудочков погибают 6,5% пациентов; к 10 годам - 33%, к 30-40 годам - 80-87%.
После проведения хирургической коррекции аномалии Эбштейна прогноз в отношении жизни становится благоприятным. Отрицательно сказываются на отдаленных результатах вмешательства выраженная кардиомегалия и развитие послеоперационной аритмии.
Врожденные пороки сердца
Врожденные пороки сердца - группа заболеваний, объединенных наличием анатомических дефектов сердца, его клапанного аппарата или сосудов, возникших во внутриутробном периоде, приводящих к изменению внутрисердечной и системной гемодинамики. Проявления врожденного порока сердца зависят от его вида; к наиболее характерным симптомам относятся бледность или синюшность кожных покровов, шумы в сердце, отставание в физическом развитии, признаки дыхательной и сердечной недостаточности. При подозрении на врожденный порок сердца выполняется ЭКГ, ФКГ, рентгенография, ЭхоКГ, катетеризация сердца и аортография, кардиография, МРТ сердца и т. д. Чаще всего при врожденных пороках сердца прибегают к кардиохирургической операции - оперативной коррекции выявленной аномалии.
Врожденные пороки сердца - весьма обширная и разнородная группа заболеваний сердца и крупных сосудов, сопровождающихся изменением кровотока, перегрузкой и недостаточностью сердца. Частота встречаемости врожденных пороков сердца высока и, по оценке различных авторов, колеблется от 0,8 до 1,2% среди всех новорожденных. Врожденные пороки сердца составляют 10-30% всех врожденных аномалий. В группу врожденных пороков сердца входят как относительно легкие нарушения развития сердца и сосудов, так и тяжелые формы патологии сердца, несовместимые с жизнью.
Многие виды врожденных пороков сердца встречаются не только изолированно, но и в различных сочетаниях друг с другом, что значительно утяжеляет структуру дефекта. Примерно в трети случаев аномалии сердца сочетаются с внесердечными врожденными пороками ЦНС, опорно-двигательного аппарата, ЖКТ, мочеполовой системы и пр.
К наиболее частым вариантам врожденных пороков сердца, встречающимся в кардиологии, относятся дефекты межжелудочковой перегородки (ДМЖП - 20%), дефекты межпредсердной перегородки (ДМПП), стеноз аорты, коарктация аорты, открытый артериальный проток (ОАП), транспозиция крупных магистральных сосудов (ТКС), стеноз легочной артерии (10-15% каждый).
Причины врожденных пороков сердца
Этиология врожденных пороков сердца может быть обусловлена хромосомными нарушениями (5%), генной мутацией (2-3%), влиянием факторов среды (1-2%), полигенно-мультифакториальной предрасположенностью (90%).
Различного рода хромосомные аберрации приводят к количественным и структурным изменениям хромосом. При хромосомных перестройках отмечаются множественные полисистемные аномалии развития, включая врожденные пороки сердца. В случае трисомии аутосом наиболее частыми пороками сердца оказываются дефекты межпредсердной или межжелудочковой перегородок, а также их сочетание; при аномалиях половых хромосом врожденные пороки сердца встречаются реже и представлены, главным образом, коарктацией аорты или дефектом межжелудочковой перегородки.
Врожденные пороки сердца, обусловленные мутациями единичных генов, также в большинстве случаев сочетаются с аномалиями других внутренних органов. В этих случаях сердечные пороки являются частью аутосомно-доминантных (синдромы Марфана, Холта-Орама, Крузона, Нунана и др.), аутосомно-рецессивных синдромов (синдром Картагенера, Карпентера, Робертса, Гурлер и др.) или синдромов, сцепленных с Х-хромосомой (синдромы Гольтца, Аазе, Гунтера и др.).
Среди повреждающих факторов внешней среды к развитию врожденных пороков сердца приводят вирусные заболевания беременной, ионизирующая радиация, некоторые лекарственные препараты, пагубные привычки матери, производственные вредности. Критическим периодом неблагоприятного воздействия на плод являются первые 3 месяца беременности, когда происходит фетальный органогенез.
Внутриутробное поражение плода вирусом краснухи наиболее часто вызывает триаду аномалий - глаукому или катаракту, глухоту, врожденные пороки сердца (тетраду Фалло, транспозицию магистральных сосудов, открытый артериальный проток, общий артериальный ствол, клапанные пороки, стеноз легочной артерии, ДМЖП и др.). Также обычно имеют место микроцефалия, нарушение развития костей черепа и скелета, отставание в умственном и физическом развитии.
В структуру эмбриофетального алкогольного синдрома обычно входят дефекты межжелудочковой и межпредсердной перегородки, открытый артериальный проток. Доказано, что тератогенное действие на сердечно-сосудистую систему плода оказывает прием амфетаминов, приводящий к транспозиции магистральных сосудов и ДМЖП; противосудорожных средств, обусловливающих развитие стеноза аорты и легочной артерии, коарктации аорты, открытого артериального протока, тетрады Фалло, гипоплазии левых отделов сердца; препаратов лития, приводящих к атрезии трехстворчатого клапана, аномалии Эбштейна, ДМПП; прогестагенов, вызывающих тетраду Фалло, другие сложные врожденные пороки сердца.
У женщин, страдающих преддиабетом или диабетом, дети с врожденными пороками сердца рождаются чаще, чем у здоровых матерей. В этом случае у плода обычно формируются ДМЖП или транспозиция крупных сосудов. Вероятность рождения ребенка с врожденным пороком сердца у женщины с ревматизмом составляет 25 %.
Кроме непосредственных причин, выделяют факторы риска формирования аномалий сердца у плода. К ним относят возраст беременной младше 15-17 лет и старше 40 лет, токсикозы I триместра, угрозу самопроизвольного прерывания беременности, эндокринные нарушения у матери, случаи мертворождения в анамнезе, наличие в семье других детей и близких родственников с врожденными пороками сердца.
Классификация врожденных пороков сердца
Существует несколько вариантов классификаций врожденных пороков сердца, в основу которых положен принцип изменения гемодинамики. С учетом влияния порока на легочный кровоток выделяют:
- врожденные пороки сердца с неизмененным (либо незначительно измененным) кровотоком в малом круге кровообращения: атрезия аортального клапана, стеноз аорты, недостаточность легочного клапана, митральные пороки (недостаточность и стеноз клапана), коарктация аорты взрослого типа, трехпредсердное сердце и др.
- врожденные пороки сердца с увеличенным кровотоком в легких: не приводящие к развитию раннего цианоза (открытый артериальный проток, ДМПП, ДМЖП, аортолегочный свищ, коарктация аорты детского типа, синдром Лютамбаше), приводящие к развитию цианоза (атрезия трехстворчатого клапана с большим ДМЖП, открытый артериальный проток с легочной гипертензией)
- врожденные пороки сердца с обедненным кровотоком в легких: не приводящие к развитию цианоза (изолированный стеноз легочной артерии), приводящие к развитию цианоза (сложные пороки сердца - болезнь Фалло, гипоплазия правого желудочка, аномалия Эбштейна)
- комбинированные врожденные пороки сердца, при которых нарушаются анатомические взаимоотношения между крупными сосудами и различными отделами сердца: транспозиция магистральных артерий, общий артериальный ствол, аномалия Тауссиг-Бинга, отхождение аорты и легочного ствола из одного желудочка и пр.
В практической кардиологии используется деление врожденных пороков сердца на 3 группы: пороки «синего» (цианотического) типа с веноартериальным шунтом (триада Фалло, тетрада Фалло, транспозиция магистральных сосудов, атрезия трехстворчатого клапана); пороки «бледного» типа с артериовенозным сбросом (септальные дефекты, открытый артериальный проток); пороки с препятствием на пути выброса крови из желудочков (стенозы аорты и легочной артерии, коарктация аорты).
Нарушения гемодинамики при врожденных пороках сердца
В результате выше названных причин у развивающего плода может нарушаться правильное формирование структур сердца, что выражается в неполном или несвоевременном закрытии перепонок между желудочками и предсердиями, неправильном образовании клапанов, недостаточном повороте первичной сердечной трубки и недоразвитии желудочков, аномальном расположении сосудов и т. д. После рождения у части детей остаются открытыми артериальный проток и овальное окно, которые во внутриутробном периоде функционируют в физиологическом порядке.
При врожденных пороках сердца бледного типа с артериовенозным сбросом вследствие гиперволемии развивается гипертензия малого круга кровообращения; при пороках синего типа с веноартериальным шунтом у больных имеет место гипоксемия.
Около 50% детей с большим сбросом крови в малый круг кровообращения погибают без кардиохирургической помощи на первом году жизни от явлений сердечной недостаточности. У детей, перешагнувших этот критический рубеж, сброс крови в малый круг уменьшается, самочувствие стабилизируется, однако постепенно прогрессируют склеротические процессы в сосудах легких, обусловливая легочную гипертензию.
При цианотических врожденных пороках сердца венозный сброс крови или ее смешение приводит к перегрузке большого и гиповолемии малого круга кровообращения, вызывая снижение насыщения крови кислородом (гипоксемию) и появление синюшности кожи и слизистых. Для улучшения вентиляции и перфузии органов развивается коллатеральная сеть кровообращения, поэтому, несмотря на выраженные нарушения гемодинамики, состояние больного может длительное время оставаться удовлетворительным. По мере истощения компенсаторных механизмов, вследствие длительной гиперфункции миокарда, развиваются тяжелые необратимые дистрофические изменения в сердечной мышце. При цианотических врожденных пороках сердца оперативное вмешательство показано уже в раннем детском возрасте.
Симптомы врожденных пороков сердца
Клинические проявления и течение врожденных пороков сердца определяется видом аномалии, характером нарушений гемодинамики и сроками развития декомпенсации кровообращения.
У новорожденных с цианотическими врожденными пороками сердца отмечается цианоз (синюшность) кожных покровов и слизистых оболочек. Синюшность усиливается при малейшем напряжении: сосании, плаче ребенка. Белые пороки сердца проявляются побледнением кожи, похолоданием конечностей.
Дети с врожденными пороками сердца обычно беспокойные, отказываются от груди, быстро устают в процессе кормления. У них появляется потливость, тахикардия, аритмии, одышка, набухание и пульсация сосудов шеи. При хроническом нарушении кровообращения дети отстают в прибавлении веса, росте и физическом развитии. При врожденных пороках сердца обычно сразу поле рождения выслушиваются сердечные шумы. В дальнейшем обнаруживаются признаки сердечной недостаточности (отеки, кардиомегалия, кардиогенная гипотрофия, гепатомегалия и др.).
Осложнениями врожденных пороков сердца могут стать бактериальный эндокардит, полицитемия, тромбозы периферических сосудов и тромбоэмболии сосудов головного мозга, застойные пневмонии, синкопальные состояния, одышечно-цианотические приступы, стенокардитический синдром или инфаркт миокарда.
Диагностика врожденных пороков сердца
Выявление врожденных пороков сердца осуществляется путем комплексного обследования. При осмотре ребенка отмечают окраску кожных покровов: наличие или отсутствие цианоза, его характер (периферический, генерализованный). При аускультации сердца нередко выявляется изменение (ослабление, усиление или расщепление) сердечных тонов, наличие шумов и пр. Физикальное обследование при подозрении на врожденный порок сердца дополняется инструментальной диагностикой - электрокардиографией (ЭКГ), фонокардиографией (ФКГ), рентгенографией органов грудной клетки, эхокардиографией (ЭхоКГ).
ЭКГ позволяет выявить гипертрофию различных отделов сердца, патологическое отклонение ЭОС, наличие аритмий и нарушений проводимости, что в совокупности с данными других методов клинического обследования позволяет судить о тяжести врожденного порока сердца. С помощью суточного холтеровского ЭКГ-мониторирования обнаруживаются скрытые нарушения ритма и проводимости. Посредством ФКГ более тщательно и детально оценивается характер, длительность и локализация сердечных тонов и шумов. Данные рентгенографии органов грудной клетки дополняют предыдущие методы за счет оценки состояния малого круга кровообращения, расположения, формы и размеров сердца, изменений со стороны других органов (легких, плевры, позвоночника). При проведении ЭхоКГ визуализируются анатомические дефекты перегородок и клапанов сердца, расположение магистральных сосудов, оценивается сократительная способность миокарда.
При сложных врожденных пороках сердца, а также сопутствующей легочной гипертензии, с целью точной анатомической и гемодинамической диагностики, возникает необходимость в выполнении зондирования полостей сердца и ангиокардиографии.
Лечение врожденных пороков сердца
Наиболее сложной проблемой в детской кардиологии является хирургическое лечение врожденных пороков сердца у детей первого года жизни. Большинство операций в раннем детском возрасте выполняется по поводу цианотических врожденных пороков сердца. При отсутствии у новорожденного признаков сердечной недостаточности, умеренной выраженности цианоза операция может быть отложена. Наблюдение за детьми с врожденными пороками сердца осуществляют кардиолог и кардиохирург.
Специфическое лечение в каждом конкретном случае зависит от разновидности и степени тяжести врожденного порока сердца. Операции при врожденных дефектах перегородок сердца (ДМЖП, ДМПП) могут включать пластику или ушивание перегородки, рентгенэндоваскулярную окклюзию дефекта. При наличии выраженной гипоксемии детям с врожденными пороками сердца первым этапом выполняется паллиативное вмешательство, предполагающее наложение различного рода межсистемных анастомозов. Подобная тактика улучшает оксигенацию крови, уменьшает риск осложнений, позволяет провести радикальную коррекцию в более благоприятных условиях. При аортальных пороках выполняется резекция или баллонная дилатация коарктации аорты, пластика аортального стеноза и др. При ОАП производится его перевязка. Лечение стеноза легочной артерии заключается в проведении открытой или эндоваскулярной вальвулопластики и т. д.
Анатомически сложные врожденные пороки сердца, при которых радикальная операция не представляется возможной, требуют выполнения гемодинамической коррекции, т. е. разделения артериального и венозного потоков крови без устранения анатомического дефекта. В этих случаях могут проводиться операции Фонтена, Сеннинга, Мастарда и др. Серьезные пороки, не поддающиеся оперативному лечению, требуют проведения пересадки сердца.
Консервативное лечение врожденных пороков сердца может включать в себя симптоматическую терапию одышечно-цианотических приступов, острой левожелудочковой недостаточности (сердечной астмы, отека легких), хронической сердечной недостаточности, ишемии миокарда, аритмий.
Прогноз и профилактика врожденных пороков сердца
В структуре смертности новорожденных врожденные пороки сердца занимают первое место. Без оказания квалифицированной кардиохирургической помощи в течение первого года жизни погибает 50-75% детей. В периоде компенсации (2-3 года) смертность снижается до 5%. Ранее выявление и коррекция врожденного порока сердца позволяет существенно улучшить прогноз.
Профилактика врожденных пороков сердца требует тщательного планирования беременности, исключения воздействия неблагоприятных факторов на плод, проведения медико-генетического консультирования и разъяснительной работы среди женщин групп риска по рождению детей с сердечной патологией, решения вопроса о пренатальной диагностике порока (УЗИ, биопсия хориона, амниоцентез) и показаниях к прерыванию беременности. Ведение беременности у женщин с врожденными пороками сердца требует повышенного внимания со стороны акушера-гинеколога и кардиолога.
Аномалия Эбштейна

Аномалия Эбштейна - редкая и тяжелая форма врожденного порока сердца. Мальформация трикуспидального клапана (ТК), обусловленная сочетанием морфологических сердечных дефектов, еще во внутриутробном периоде приводит к развитию тахикардии и выраженной кадиомегалии. И только своевременное обнаружение и радикальная коррекция реально улучшают жизненный прогноз.
Рассказывает специалист ЦМРТ
Дата публикации: 02 Августа 2021 года
Дата проверки: 30 Ноября 2021 года
Содержание статьи
Причины недостаточности трехстворчатого клапана
Болезнь Эбштейна составляет порядка 1% от общего количества пренатально диагностируемых пороков. Клинически доказано, что в большинстве случаев мальформация ТК развивается на фоне приема в I-II триместре беременности препаратов лития. Также существуют тератогенные факторы, способные оказать негативное влияние на формирование сердца плода:
- алкоголь
- радиоактивное излучение
- постоянная угроза самопроизвольного аборта
- тяжелый токсикоз
- инфекционные и соматические заболевания матери
В отдельных случаях аномалия Эбштейна у детей рассматривается, как врожденный порок с наследственным компонентом. Это подтверждают семейные случаи заболевания.
Симптомы патологии трехстворчатого клапана
Дефект развития клапана, находящегося между правым предсердием и правым желудочком, обуславливается неправильным прикреплением его створок. При незначительных гемодинамических нарушениях морфо-функциональное нарушение может протекать бессимптомно. Самые характерные признаки аномалии: низкая физическая активность, прогрессирующий или выраженный диффузный цианоз (синюшность кожных покровов), боли в области сердца, периодические приступы сердцебиения.
При тяжелой форме заболевания, в 90% случаев, наступает внутриутробная гибель плода, в 80% - смерть в раннем младенчестве. При естественном течении болезни до взрослого возраста доживает только порядка 34% больных. У пациентов наблюдается одышка, набухание и пульсация шейных вен. Нередко увеличивается печень, дистальные фаланги пальцев утолщаются, приобретая вид барабанных палочек.
Ногтевые пластины деформируются (симптом «часовых стекол), средний отдел грудной клетки выпячивается, образуя «сердечный горб». Тяжелые нарушения ритма, кардиологический синдром «бычьего сердца» и прогрессирующая сердечная недостаточность, при отсутствии необходимого лечения, становятся причиной летального исхода.
Классификация и стадии аномалии Эбштейна
Врожденный порок ТК имеет 5 анатомических вариантов:
- Тип А: изменения минимальны, объем движений не лимитирован
- Тип В: передняя створка утолщена и подвижна, септальная и задняя смещены или полностью отсутствуют
- Тип С: отверстие 3-хстворчатого клапана сглажено и малофункционально, створки существенно уменьшены и сдвинуты к верхушке сердца
- Тип D: передняя створка образует конгломерат с модераторным пучком, задняя и септальная недоразвиты
- Тип Е: все створки срастаются и, вместе с приточным отделом, образуют единый трехстворчатый мешок. Миокард полностью истончен, сокращение практически отсутствует
На I стадии болезни Эбштейна не проявляется характерных симптомов. На II стадии, сопровождающейся выраженными расстройствами гемодинамики, врожденный порок может протекать без нарушений ритма или сопровождаться аритмией. III стадия - декомпенсированная (терминальная).
Методы диагностики
Пациентам с подозрением на недостаточность трехстворчатого клапана назначается комплекс диагностических мероприятий:
- физикальное обследование (аускультация)
- тесты с физической нагрузкой
- лабораторные анализы крови (определение уровня гемоглобина и насыщенности кислородом)
- ЭКГ
- рентгенография грудной клетки
- УЗИ сердца
- МРТ
- КТ
- ЭФИ (электрофизиологическое исследование проводящей системы).
Дополнительно, для уточнения типа и степени тяжести заболевания, в кардиологии применяется катетеризация полостей сердца и апекскардиография.
К какому врачу обратиться
Ведение пациентов с врожденными пороками сердца занимается кардиолог и кардиохирург.
Как лечить патологию
Лечение аномалии Эбштейна может быть консервативным и оперативным. Медикаментозная терапия признана симптоматической методикой, направленной на облегчение состояния и коррекцию осложнений болезни I-II функционального класса. Она включает антикоагулянты, диуретики и антиаритмические препараты.
При прогрессирующем увеличении и дисфункции правого желудочка, развитии сердечной недостаточности и повышении риска внезапной смерти методом выбора становится кардиохирургическая операция. Для восстановления запирательной функции и нормализации гемодинамики проводится пластика или протезирование трикуспидального клапана. Одномоментно устраняются имеющиеся сопутствующие пороки и дефекты межпредсердной перегородки.
Последствия
Отсутствие необходимого лечения врожденной аномалии влечет за собой ряд опасных для жизни осложнений:
- аритмии
- кардиомиопатия (патаология сердечной мышцы)
- тяжелая кардиомегалия (увеличение размеров и массы сердца)
- сердечная недостаточность
- инсульт
- летальный исход
Профилактика
Чтобы защитить будущего ребенка и минимизировать риск развития ВПС, беременным женщинам настоятельно рекомендуется проводить своевременное лечение имеющихся инфекционно-воспалительных и соматических заболеваний. В период вынашивания плода необходимо выполнять все врачебные рекомендации, исключить прием препаратов на основе лития и любые токсичные тератогенные воздействия.
Лечение аномалии Эбштейна в клиниках ЦМРТ
Сеть клиник ЦМРТ в Санкт-Петербурге предлагает комплексные программы восстановительного лечения пациентов с врожденными пороками сердца. Индивидуальные поэтапные курсы включают:
- подбор и коррекцию медикаментозной терапии
- повышение толерантности к физическим нагрузкам (под контролем реабилитолога)
- модификацию факторов риска
- физиотерапевтическое воздействие
Желающим записаться на консультативный прием к кардиологу необходимо оставить заявку на сайте ЦМРТ или позвонить.
Синдром WPW
Синдром Вольфа-Паркинсона-Уайта (синдром WPW) - клинико-электрокардиографический синдром, характеризующийся предвозбуждением желудочков по дополнительным атриовентрикулярным путям проведения и развитием пароксизмальных тахиаритмий. Синдром WPW сопровождается различными аритмиями: наджелудочковой тахикардией, фибрилляцией или трепетанием предсердий, предсердной и желудочковой экстрасистолией с соответствующей субъективной симптоматикой (ощущением сердцебиения, одышкой, гипотензией, головокружением, обмороками, болями в грудной клетке). Диагностика синдрома WPW основана на данных ЭКГ, суточного ЭКГ-мониторирования, ЭхоКГ, ЧПЭКС, ЭФИ. Лечение синдрома WPW может включать антиаритмическую терапию, чреспищеводную электрокардиостимуляцию, катетерную РЧА.
Синдром Вольфа-Паркинсона-Уайта (синдром WPW) - синдром преждевременного возбуждения желудочков, обусловленный проведением импульсов по добавочным аномальным проводящим пучкам, соединяющим предсердия и желудочки. Распространенность синдрома WPW, по данным кардиологии, составляет 0,15-2%. Синдром WPW чаще встречается среди мужчин; в большинстве случаев манифестирует в молодом возрасте (10-20 лет), реже - у лиц старшего возраста. Клиническое значение синдрома WPW заключается в том, что при его наличии часто развиваются тяжелые нарушения сердечного ритма, которые представляют угрозу для жизни больного и требуют особых подходов к лечению.
Причины синдрома WPW
По мнению большинства авторов, синдром WPW, обусловлен сохранением добавочных атриовентрикулярных соединений в результате незавершенного кардиогенеза. При этом происходит неполная регрессия мышечных волокон на этапе формирования фиброзных колец трикуспидального и митрального клапанов.
В норме дополнительные мышечные пути, соединяющие предсердия и желудочки, существуют у всех эмбрионов на ранних стадиях развития, но постепенно они истончаются, сокращаются и полностью исчезают после 20-й недели развития. При нарушении формирования фиброзных атриовентрикулярных колец мышечные волокна сохраняются и составляют анатомическую основу синдрома WPW. Несмотря на врожденный характер дополнительных АВ-соединений, синдром WPW может впервые проявиться в любом возрасте. При семейной форме синдрома WPW чаще имеют место множественные добавочные атриовентрикулярные соединения.
Классификация синдрома WPW
По рекомендации ВОЗ, различают феномен и синдром WPW. Феномен WPW характеризуется электрокардиографическими признаками проведения импульса по дополнительным соединениям и предвозбуждением желудочков, но без клинических проявлений АВ реципрокной тахикардии (re-entry). Под синдромом WPW подразумевается сочетание предвозбуждения желудочков с симптоматической тахикардией.
С учетом морфологического субстрата выделяют несколько анатомических вариантов синдрома WPW.
I. С добавочными мышечными АВ-волокнами:
- идущими через добавочное левое или правое париетальное АВ-соединение
- идущими через аортально-митральное фиброзное соединение
- идущими от ушка правого или левого предсердия
- связанными с аневризмой синуса Вальсальвы или средней вены сердца
- септальными, парасептальными верхними или нижними
II. Со специализированными мышечными АВ-волокнами («пучками Кента»), происходящими из рудиментарной, аналогичной структуре атриовентрикулярного узла, ткани:
- атрио-фасцикулярными - входящими в правую ножку пучка Гиса
- входящими в миокард правого желудочка.
Выделяют несколько клинических форм синдрома WPW:
- а) манифестирующую - с постоянным наличием дельта-волны, синусовым ритмом и эпизодами атриовентрикулярной реципрокной тахикардии.
- б) интермиттирующую - с преходящим предвозбуждением желудочков, синусовым ритмом и верифицированной атриовентрикулярной реципрокной тахикардией.
- в) скрытую - с ретроградным проведением по дополнительному атриовентрикулярному соединению. Электрокардиографические признаки синдрома WPW не выявляются, имеются эпизоды атриовентрикулярной реципрокной тахикардии.
Патогенез синдрома WPW
Синдром WPW обусловлен распространением возбуждения от предсердий к желудочкам по дополнительным аномальным путям проведения. В результате этого возбуждение части или всего миокарда желудочков происходит раньше, чем при распространении импульса обычным путем - по АВ-узлу, пучку и ветвям Гиса. Предвозбуждение желудочков отражается на электрокардиограмме в виде дополнительной волны деполяризации - дельта-волны. Интервал P-Q(R) при этом укорачивается, а длительность QRS увеличивается.
Когда в желудочки приходит основная волна деполяризации, их столкновение в сердечной мышце регистрируется в виде так называемого сливного комплекса QRS, который становится несколько деформированным и уширенным. Нетипичное возбуждение желудочков сопровождается нарушением последовательности процессов реполяризации, что находит выражение на ЭКГ в виде дискордантного комплексу QRS смещения RS-T сегмента и изменения полярности зубца T.
Возникновение при синдроме WPW пароксизмов суправентрикулярной тахикардии, мерцания и трепетания предсердий связано с формированием круговой волны возбуждения (re-entry). В этом случае импульс по AB-узлу движется в антероградном направлении (от предсердий к желудочкам), а по дополнительным путям - в ретроградном направлении (от желудочков к предсердиям).
Симптомы синдрома WPW
Клиническая манифестация синдрома WPW происходит в любом возрасте, до этого его течение может быть асимптомным. Синдром WPW сопровождается различными нарушениями сердечного ритма: реципрокной наджелудочковой тахикардией (80%), фибрилляцией предсердий (15-30%), трепетанием предсердий (5%) с частотой 280-320 уд. в мин. Иногда при синдроме WPW развиваются менее специфичные аритмии - предсердная и желудочковая экстрасистолия, желудочковая тахикардия.
Приступы аритмии могут возникать под влиянием эмоционального или физического перенапряжения, злоупотребления алкоголем или спонтанно, без видимых причин. Во время аритмического приступа появляются ощущения сердцебиения и замирания сердца, кардиалгии, чувство нехватки воздуха. Мерцание и трепетание предсердий сопровождается головокружением, обмороками, одышкой, артериальной гипотензией; при переходе в фибрилляцию желудочков может наступить внезапная сердечная смерть.
Пароксизмы аритмии при синдроме WPW могут длиться от нескольких секунд до нескольких часов; иногда они купируются самостоятельно или после выполнения рефлекторных приемов. Затяжные пароксизмы требуют госпитализации больного и вмешательства кардиолога.
Диагностика синдрома WPW
При подозрении на синдром WPW проводится комплексная клинико-иснтрументальная диагностика: ЭКГ в 12 отведениях, трансторакальная эхокардиография, мониторирование ЭКГ по Холтеру, чреспищеводная электрокардиостимуляция, электрофизиологическое исследование сердца.
К электрокардиографическим критериям синдрома WPW относятся: укорочение PQ-интервала (менее 0,12 с), деформированный сливной QRS-комплекс, наличие дельта-волны. Суточное ЭКГ мониторирование применяется для обнаружения преходящих нарушений ритма. При проведении УЗИ сердца выявляются сопутствующие пороки сердца, кардиомиопатию.
Проведение чреспищеводной электрокардиостимуляции при синдроме WPW позволяет доказать наличие дополнительных путей проведения, индуцировать пароксизмы аритмии. Эндокардиальное ЭФИ позволяет точно определить локализацию и количество дополнительных путей, верифицировать клиническую форму синдрома WPW, выбрать и оценить эффективность лекарственной терапии или РЧА. Дифференциальную диагностику синдрома WPW проводят с блокадами ножек пучка Гиса.
Лечение синдрома WPW
При отсутствии пароксизмов аритмии синдром WPW не требует специального лечения. При гемодинамически значимых приступах, сопровождающихся синкопэ, стенокардией, гипотензией, нарастанием признаков сердечной недостаточности, требуется выполнение незамедлительной наружной электрической кардиоверсии или чреспищеводной электрокардиостимуляции.
В некоторых случаях для купирования пароксизмов аритмий эффективными оказываются рефлекторные вагусные маневры (массаж каротидного синуса, проба Вальсальвы), внутривенное введение АТФ или блокаторов кальциевых каналов (верапамила), антиаритмических препаратов (новокаинамида, аймалина, пропафенона, амиодарона). В дальнейшем пациентам с синдромом WPW показана постоянная антиаритмическая терапия.
В случае резистентности к антиаритмическим препаратам, развития фибрилляцией предсердий проводится катетерная радиочастотная абляция добавочных путей проведения трансаортальным (ретроградным) или транссептальным доступом. Эффективность РЧА при синдроме WPW достигает 95%, риск рецидивов составляет 5-8 %.
Прогноз и профилактика синдрома WPW
У пациентов с бессимптомным течением синдрома WPW прогноз благоприятный. Лечение и наблюдение требуется только лицам, имеющим отягощенный семейный анамнез в отношении внезапной смерти и профессиональные показания (спортсменам, летчикам и др.). При наличии жалоб или жизнеугрожающих аритмий необходимо проведение полного комплекса диагностического обследования для выбора оптимального метода лечения.
Пациенты с синдромом WPW (в том числе, перенесшие РЧА) нуждаются в наблюдении кардиолога-аритмолога и кардиохирурга. Профилактика синдрома WPW носит вторичный характер и заключается в проведении противоаритмической терапии для предотвращения повторных эпизодов аритмий.
Вирус Эпштейна-Барр
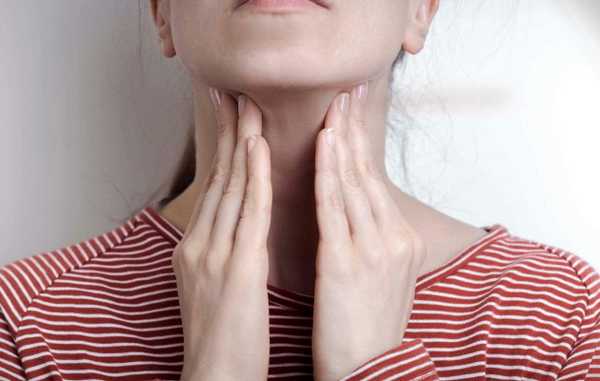
ВЭБ вирус, или вирус Эпштейна-Барра - это вирус герпеса 4 типа, который вызывает высококонтагиозное инфекционное заболевание у взрослых и детей. По данным ВОЗ, вирус персистирует в организме 75-90% взрослого и детского населения. До 5% случаев заканчивается малигнизацией новообразований.
Дата публикации: 25 Августа 2021 года
Причины Эпштейн-Барра
Вирус Эпштейна-Барра относится к группе герпесвирусов 4 типа.
В отличие от простого герпеса Эпштейн Барр приводит к пролиферации, или патологическому разрастанию, пораженных тканей с развитием образований. Вирус персистирует не в нервных клетках, а в клетках иммунной системы. Еще одно отличие - его постоянная трансформация.
Передаваться вирус Эпштейн Барр может:
- трансплацентарно
- контактно-бытовым путем
- половым способом
- воздушно-капельным путем
Заражение происходит только в острой стадии заболевания.
После перенесенной инфекции человек в течение 2 лет может оставаться возможным источником заражения. До 30% переболевших людей остаются вирусоносителями.
При воздушно-капельном типе заражения после выделения больным человеком ВЭБ попадает на слизистые оболочки дыхательной системы здоровых людей. Далее с током лимфы вирус попадает в организм.
Протекать болезнь может бессимптомно или по типу простуды. Острая фаза встречается при снижении иммунного ответа. Чаще развивается хронический процесс.
Симптомы Эпшетйна Барра
Начальные симптомы Эпштейн Барра в острой стадии схожи с простудой. Заболевание носит название «Инфекционный мононуклеоз». Инкубационный период колеблется от 2 до 60 дней, в среднем занимает 2-20 дней.
Ведущими признаками инфекции являются:
Симптомами вируса Эпштейна Барр у взрослых могут быть:
- слабость
- головокружение
- снижение работоспособности
- высокая температура
- боли в горле, насморк, кашель
- тяжесть в правом подреберье
- потемнение мочи
- желтушность кожного покрова и слизистой глаз
Характерные признаки заболевания длятся в среднем 7-14 дней, после чего наступает выздоровление. Слабость и увеличенные лимфоузлы сохраняются до 21 дня.
Для хронического течения заболевания характерно появление новообразований различной локализации.
Стадии развития болезни Эпштейн Барра
В стадии развития болезни Эпштейн Барра выделяют фазы: острую, или инфекционный мононуклеоз и хроническую.
Острая фаза занимает до 3 недель. Далее заболевание переходит в хронический процесс, который протекает с периодами обострения и ремиссий.
По тяжести течения выделяют 3 степени: легкая, средняя, тяжелая.
Эпштейн Барр при беременности
При первичном заражении вирусом Эпштейна Барр во время беременности могут развиваться осложнения со стороны матери и плода:
- самопроизвольный выкидыш
- преждевременные роды
- внутриутробное заражение плода
- поражение печени и селезенки матери.
Если будущая мать ранее имела контакт с возбудителем, то возможен переход инфекции из латентного состояния в период обострения.
Лечение инфекционного мононуклеоза у беременной обязательно проводится в стационаре под наблюдением специалистов.
Как диагностировать
Диагностика болезни Эпштейн Барра комплексная и состоит из сбора жалоб, осмотра и анализов.
При обращении пациентки к врачу отмечается характерная для простудных заболеваний симптоматика. При этом развивается регионарная лимфаденопатия и гепатоспленомегалия.
При осмотре определяются увеличенные до 2 см подвижные умеренно болезненные лимфоузлы. Поражаются обычно несколько групп лимфоузлов. При шейной лимаденопатии может возникать одутловатость лица.
Назначается общий анализ крови, для которого характерно повышение числа лейкоцитов, лимфоцитов и моноцитов, ускоренное СОЭ
В биохимическом анализе отмечается повышение значений печеночных ферментов: АлАТ, АсАТ, ЛДГ, билирубина.
Для верификации возбудителя используется серологическая диагностика:
- ПЦР: позволяет определить ДНК вируса
- ИФА: определяются количество и класс иммуноглобулинов, или антитела к Эпштейн Барру
- Для определения степени поражения печени и селезенки назначается УЗИ органов брюшной полости
- Дополнительно может назначаться иммунологическое исследование при тяжелых формах болезни.
Лечением острой вирусной инфекции Эпштейн Барр занимается врач-инфекционист, терапевт или ВОП. Хроническую форму с развитием новообразований лечит онколог. При наличии показаний пациент направляется к смежным специалистам: ЛОР, иммунолог, гематолог.
Как лечить болезнь Эпштейн Барр
Лечение неспецифическое. Инфекционный мононуклеоз необходимо лечить в стационарных условиях.
Для снижения вирусной активности назначаются противовирусные препараты. По показаниям назначаются антибиотики, иммуномодуляторы.
Обязательно проводится симптоматическая терапия:
- при температуре назначаются жаропонижающие
- при кашле - отхаркивающие
- при заложенности носа - сосудосуживающие капли
Лечение патологии занимает от 2 недель до нескольких месяцев.
Вирус Эпштейн Барра может приводить к развитию осложнений в виде:
- отита
- дыхательной недостаточности
- гепатита
- гемолитической анемии
- тромбоцитопенической пурпуры
- панкреатита
- малигнизации опухолей
Специфической профилактики заболевания не существует. Проводятся мероприятия по защите слизистых оболочек от проникновения вируса от больного человека: ношение маски при наличии симптомов простудной болезни, своевременное обращение к врачу для определения тактики ведения и лечения.
Лечение болезни Эпштейн Барра в клиниках ЦМРТ
В клинике ЦМРТ ведет прием терапевт. Специалист назначает необходимые виды лабораторно-диагностических исследований. При наличии жалоб можно записаться на прием по телефону, через онлайн-форму на сайте.
Источники
Инфекционные болезни: Уч. Пос / И.А. Бережнова. - М.: Риор, 2015.
Инфекционные болезни / Б.П. Богомолов. - М.: МГУ, 2006
Вирусные инфекционные заболевания и их этиотропная терапия: Метод. рек. для врачей, клин. ординаторов, интернов, студ. мед. вузов. / В. Л. Кокорев, Н. П. Куприна, Л. М. Коноплина и др.; Воронеж.мед.акад.;Сост.С.П.Кокорева и др. - Воронеж, 2003.
Читайте также:
- Острые респираторные заболевания. Диагностика причины респираторного заболевания
- Даназол. Избыток андрогенов у женщин и гирсутизм
- Обмороки у больных раком головы и шеи. Паранеопластические синдромы.
- Группы бронхэктатической болезни. Клиническое течение бронхэктазий у пациенки
- Предполагаемый срок родов. Как определяют предполагаемый срок родов?