Болезнь минимальных изменений
Добавил пользователь Алексей Ф. Обновлено: 01.02.2026
В данных клинических рекомендациях новые и узконаправленные термины не используются.
1. Краткая информация
1.1. Определение
- клинико-лабораторный симптомокомплекс, характеризующийся протеинурией (>50 мг/кг/сут или > 40 мг/м 2 /час, т.е. 2,5 г/сут и более), гипоальбуминемией (
1.2. Этиология и патогенез
Нефротический синдром может быть первичным (при изолированном поражении почек) и вторичным (в составе системных заболеваний, на фоне инфекций). Выделяют также нефротический синдром, связанный с генетической патологией. Основной механизм возникновения - увеличение проницаемости клубочкового фильтра для белка вследствие функционального или структурного повреждения. В результате потери белка с мочой развивается гипопротеинемия и гипоальбуминемия, ведущие к падению онкотического давления плазмы с развитием отеков. Нефротический синдром у большинства детей является идиопатическим.
Патогенез протеинурии при идиопатическом нефротическом синдроме с минимальными изменениями, наиболее часто встречающимся у детей, до конца не изучен. Наиболее признана гипотеза о повышении гломерулярной проницаемости для белков плазмы в результате воздействия циркулирующих факторов на капилляры гломерул и повреждение «щелевых» диафрагм между отростками подоцитов. Предполагается, что активированные Т-лимфоциты продуцируют лимфокины, которые влияют на проницаемость гломерул для белков плазмы и вызывают протеинурию. [1],[2],[3],[4]
1.3. Эпидемиология
Ежегодная частота возникновения нефротического синдрома составляет 2-7 первичных случаев на 100 000 детского населения, распространённость у детей - 12-16 случаев на 100 000 детской популяции [2].
1.4. Кодирование по МКБ-10
Нефротический синдром (N04):
- Нефротический синдром с незначительными гломерулярными нарушениями
- Нефротический синдром при очаговых и сегментарных гломерулярных повреждениях
- Нефротический синдром при диффузном мембранозном гломерулонефрите
- Нефротический синдром при диффузном мезангиальном пролиферативном гломерулонефрите
- Нефротический синдром при диффузном эндокапиллярном пролиферативном гломерулонефрите
- Нефротический синдром при диффузном мезангиокапиллярном гломерулонефрите
- Нефротический синдром при болезни плотного осадка
- Нефротический синдром при диффузном серповидном гломерулонефрите
- Нефротический синдром с другими изменениями
- Нефротический синдром с неуточненным изменением
1.5. Примеры диагнозов
Идиопатический нефротический синдром, стероидрезистентный вариант, активная стадия. Хроническая болезнь почек 1 стадия. Морфологический диагноз: фокально-сегментарный гломерулосклероз.
Идиопатический нефротический синдром, стероидзависимый вариант, стадия клинико-лабораторной ремиссии. Хроническая болезнь почек 1 стадия. Морфологический диагноз: болезнь минимальных изменений.
Идиопатический нефротический синдром, стероидчувствительный вариант, часто рецидивирующее течение, стадия клинико-лабораторной ремиссии. Хроническая болезнь почек 1 стадия.
1.6. Классификация
НС подразделяют на идиопатический (первичный) и вторичный.
Идиопатический НС развивается при заболеваниях собственно клубочков почек.
Вторичный НС вызывается многочисленной группой различных заболеваний, которые обуславливают формирование специфической нефропатии (Наследственные заболевания (поликистоз почек, синдром Альпорта, спондилоэпифизарная дисплазия, болезнь Фабри, синдром Марфана и пр.); ревматические болезни (системная красная волчанка, системная склеродермия, дерматомиозит, ревматоидный артрит, ревматизм); системные васкулиты (геморрагический васкулит, узелковый полиартериит, гранулематоз Вегенера); гемолитико-уремический синдром; рефлюкс-нефропатия; амилоидоз почек; сахарный диабет; болезни крови (лимфогранулематоз, смешанная криоглобулинемия, миеломная болезнь, серповидно-клеточная анемия, талассемия); тромбозы вен и артерий почек, аорты или нижней полой вены; опухоли различной локализации; лекарственное поражение почек (препараты висмута, золота, противоэпилептические препараты и др.); болезни вирусной этиологии (гепатит B и C, цитомегаловирусная инфекция, ВИЧ-инфекция); болезни бактериальной этиологии (септический эндокардит; пневмония, абсцессы, бронхоэктазы, остеомиелит; туберкулёз, сифилис).
В зависимости от ответа на стандартный курс терапии преднизолоном нефротический синдром принято делить на стероидчувствительный и стероидрезистентный.
Стероидчувствительный НС - как правило, это дети с болезнью минимальных изменений (БМИ); ремиссия достигается в течение 2-4 недель, еще у части пациентов - к 6-8 неделе и только у 4% - через 12 недель от начала лечения:
— стероидчувствительный, нерецидивирующий после однократного курса стероидной терапии с достижением полной длительной ремиссии;
— стероидчувствительный, нечасто рецидивирующий - после достижения ремиссии по окончанию первого курса стероидной терапии рецидивы отмечаются реже, чем 2 раза в 6 месяцев;
— стероидчувствительный, часто рецидивирующий - после достижения ремиссии рецидивы - не реже 2 раз в 6 месяцев;
— стероидчувствительный стероидзависимый - рецидив развивается при снижении дозы преднизолона или не позднее, чем через 2 недели после отмены препарата;
2. Диагностика
2.1. Жалобы и анамнез
Жалобы на появление отеков и уменьшение количества мочи. Первым клиническим симптомом, заметным для больного и окружающих, являются отеки. Они могут развиться постепенно или же стремительно, достигнув степени анасарки. [1],[2],[3],[4].
2.2. Физикальное обследование
Периферические отеки выявляются в области век, лица, поясничной области и половых органов, могут распространяться на всю подкожную клетчатку, растягивая кожу до образования striae distensae. В это время у больных могут образовываться транссудаты в серозные полости: одно- или двусторонний гидроторакс, асцит, гидроперикард; возможно развитие отека легких.
При обследовании пациента обязательно рекомендуется измерение артериального давления, которое может быть повышено у детей с активной стадией нефротического синдрома. [1],[2],[3],[4]
(Сила рекомендации 1; уровень доказательств B)
2.3. Лабораторная диагностика
Рекомендуется определение белка в общем анализе мочи [1],[2].
(Сила рекомендации 1;уровень доказательств B)
Рекомендуется определение суточной экскреции белка с мочой [1],[2].
(Сила рекомендации 2; уровень доказательств A)
Комментарий: диагностически значимой для нефротического синдрома является протеинурия >50 мг/кг/сут или >40 мг/м 2 /сут, т.е. 2,5 г/сут и более. При невозможности определения суточной экскреции белка для уточнения степени протеинурии может быть использовано определение отношения уровня экскретируемого белка к креатинину в разовой порции мочи. Этот коэффициент достоверно коррелирует с уровнем суточной протеинурии/1,73м 2 .
Рекомендуется определение эритроцитов и лейкоцитов в анализе мочи [1],[2].
(Сила рекомендации 1; уровень доказательств D)
Комментарий: Гематурия не характерна для нефротического синдрома, но может сопровождать его, являясь признаком пролиферативных вариантов гломерулонефрита, наследственного нефрита и т.д., может быть разной степени выраженности - от умеренной до макрогематурии, лейкоцитурия также могут присутствовать у детей с нефротическим синдромом (см. раздел дифференциальный диагноз).
Рекомендовано проведение биохимического анализа крови (общий белок, альбумин, холестерин, креатинин, натрий, калий, кальций) [1],[2]:
Комментарии: Для нефротического синдрома характерны:
— гипопротеинемия: общий белок крови снижается до 40-30 г/л.
— гиперлипидемия: наиболее характерно повышение содержания в сыворотке крови холестерина, триглицеридов, а также дислипопротеинемия.
При исследовании биохимического анализа крови следует обращать внимание на уровень креатинина (может быть повышен), что является следствием гиповолемии при нефротическом синдроме, снижение уровня электролитов (гипонатриемия, гипокальциемия).
Рекомендовано исследование коагулограммы (фибриноген, уровень антитромбина III в сыворотке крови).
Комментарий: при нефротическом синдроме может повышаться уровень фибриногена, снижаться уровень антитромбина III.
Рекомендовано исследование общего анализа крови [1],[2].
Комментарий: высокая СОЭ является признаком активности нефротического синдрома и гипопротеинемии. Лейкоцитоз может быть следствием как приема кортикостероидных препаратов, так и проявлением бактериальной инфекции, которая часто осложняет течение нефротического синдрома. При почечной недостаточности может развиваться анемия.
2.4. Инструментальная диагностика
Рекомендовано измерение АД, в том числе суточное мониторирование АД, при наличии показаний [1,2].
(Сила рекомендаций 1; уровень доказательств С)
Рекомендовано проведение Эхо-КГ для оценки морфометрических параметров сердца и крупных сосудов при отеках, артериальной гипертензии, для выявления гидроперикарда [1],[2].
Рекомендовано проведение ЭКГ для выявления признаков возможных электролитных нарушений [1],[2].
Рекомендовано проведение ультразвукового исследования (УЗИ) почек (с допплерографией внутрипочечных сосудов) [1],[2].
Рекомендовано проведение денситометрии поясничного отдела позвоночника или рентгенографии трубчатых костей при длительной терапии глюкокортикостероидами для оценки степени деминерализации костной ткани [1],[2].
Рекомендовано проведение пункционной биопсии почки по показаниям с последующей световой, и, при необходимости, иммунофлюоресцентной и электронной микроскопией почечной ткани для уточнения морфологии ее повреждения [1],[2].
Комментарий: Показания к биопсии почки при нефротическом синдроме:
— стероидрезистентность нефротического синдрома (первичная и вторичная);
— НС у детей младше 1 года и старше 12 лет;
— через 2,5-3 года после начала лечения ингибиторами кальциневрина или при снижении функции почек на фоне этой терапии.
2.5. Иная диагностика
Для уточнения генеза нефротического синдрома рекомендовано назначение дополнительных лабораторных исследований [1],[2]:
вирусологические исследования: маркеры вирусов гепатита В, С (при подозрении на вторичный гломерулонефрит, связанный с хроническими гепатитами);
иммунологическое исследование крови при подозрении на системные заболевания: анти-ДНК, антинуклеарный фактор (АНФ), С3-фракция комплемента, криоглобулины;
исследование уровня Антистрептолизина-О (АСЛ-О) в крови при подозрении на острый постинфекционный гломерулонефрит,
молекулярно-генетическое исследование при стероидрезистентном нефротическом синдроме для определения мутации генов нефрина (NPHS1) и подоцина (NPHS2).
(Сила рекомендации 2; уровень доказательности B)
2.6. Дифференциальный диагноз
Проводится между гломерулопатиями, которые могут быть причиной нефротического синдрома.
Болезнь минимальных изменений (БМИ) - наиболее частая причина идиопатического нефротического синдрома у детей.
Фокально-сегментарный гломерулосклероз (ФСГС) - одна из основных форм стероидрезистентного идиопатического нефротического синдрома, составляет 10-18% случаев среди всех детей с идиопатическим нефротическим синдромом и 45% в целом в структуре стероидрезистентного нефротического синдрома. Диагноз ФСГС устанавливается по результатам биопсии почки.
Быстро-прогрессирующий гломерулонефрит морфологически характеризуется формированием полулуний более чем в 50 % клубочков. Клинически заболевание проявляется прогрессированием до конечной стадии хронической почечной недостаточности в течение от нескольких недель до нескольких месяцев.
Мембранопролиферативный (мезангиокапиллярный) гломерулонефрит (МПГН) нечастое заболевание у детей, более характерно для подросткового возраста. Нефротический синдром носит стероидрезистентный характер, в большинстве случаев сочетается с гематурий и гипокомплементемией. Выделяют 2 типа МПГН, различающиеся электронно-микроскопически и механизмом активации комплемента.
IgA-нефропатия - мезангиопролиферативный гломерулонефрит с преимущественным отложением IgA, выявляемым при иммунофлюоресцентной микроскопии. Проявляется, в основном, микрогематурией с протеинурией разной степени выраженности. Характерны эпизоды макрогематурии на фоне острых респираторных инфекций.
Мембранозная нефропатия - частая причина идиопатического нефротического синдрома у взрослых (до 50% случаев). У детей наиболее часто встречается вторичная мембранозная нефропатия при системной красной волчанке (СКВ), вирусном гепатите В, сифилисе, малярии.[1],[2],[3],[4]
Болезнь минимальных изменений

Болезнь минимальных изменений имеет острое начало и проявляется отеками и тяжелой протеинурией, главным образом у детей. Почечная функция обычно сохранена. Диагноз устанавливается эмпирически или на основании биопсии почки. Прогноз благоприятный. Лечение включает назначение глюкокортикоидов или циклофосфамида или циклоспорина для пациентов, не отвечающих на глюкокортикоидную терапию.
Болезнь минимальных изменений (БМИ) является самой частой причиной нефротического синдрома у детей 4-8 лет (80-90% нефротического синдрома у детей), но она также встречается и у взрослых (10-20% нефротического синдрома у взрослых). Причина почти всегда неизвестна, хотя в редких случаях возможно вторичное развитие болезни на фоне приема лекарственных препаратов (особенно нестероидных противовоспалительных препаратов [НПВС]) и лимфопролиферативных заболеваний (особенно лимфомы Ходжкина Лимфома Ходжкина Лимфома Ходжкина представляет собой локализованную или диссеминированную пролиферацию клеток лимфоретикулярной системы, которая может поражать лимфатические узлы, селезенку, печень или костный. Прочитайте дополнительные сведенияКлинические проявления
Болезнь минимальных изменений вызывает нефротический синдром, как правило, без артериальной гипертензии или азотемии; микрогематурия встречается приблизительно у 20% пациентов, преимущественно взрослых. Азотемия может встречаться при вторичных случаях заболевания и у больных старше 60 лет. Пациенты с минимальными изменениями заболевания теряют альбуминс мочой в большем количестве, чем более крупные сывороточные белки, вероятно, потому, что болезнь вызывает изменения заряда гломерулярного фильтрационного барьера, который влияет на альбумин избирательно.
Диагностика
У взрослых с идиопатическим нефротическим синдромом проводят биопсию почек
У детей диагноз может быть заподозрен (и начато лечение) на основе следующих типичных характеристик:
Внезапное необъяснимое появление протеинурии нефротического диапазона, в основном за счет альбумина
Нормальная функция почек
Ненефритический мочевой осадок
Биопсия почек необходима взрослым и детям с атипичными проявлениями. При электронной микроскопии определяется отек с диффузным набуханием отростков (ножек) эпителиальных подоцитов (см. рисунок Признаки иммунологической гломерулярной патологии при электронной микроскопии Признаки иммунологической гломерулярной патологии при электронной микроскопии ). При иммунофлюоресцентном исследовании не находят отложений комплемента и иммуноглобулинов. Хотя сглаживание не наблюдают в отсутствии протеинурии, тяжелая протеинурия может встречаться и при нормальных отростках подоцитов.
Признаки иммунологической гломерулярной патологии при электронной микроскопии
Лечение
Иногда циклофосфамид или циклоспорин
Кортикостероиды
Спонтанная ремиссия происходит в 40% случаев, но большинство пациентов принимают кортикостероиды. Около 80 до 90% пациентов реагируют на начальную терапию кортикостероидами (например, преднизон 60 мг/м 2 перорально 1 раз в день в течение от 4 до 6 недель у детей и от 1 до 1,5 мг/кг перорально 1 раз в день в течение от 6 до 8 недель у взрослых), но у 40-73% респондентов возникает рецидив. Пациенты, которые отвечают на терапию (например, уменьшается протеинурия или увеличивается диурез, если есть отеки), должны продолжать принимать преднизолон еще в течение 2 недель и перейти на поддерживающий режим, чтобы минимизировать побочные эффекты (2-3 мг/кг через день в течение 4-6 недель у детей и 8-12 недель у взрослых, постепенное снижение дозы в течение следующих 4 месяцев). Более длительная исходная терапия и более медленная отмена преднизолона снижают частоту рецидивов. Нечувствительность к данной терапии может быть обусловлена предсуществующим фокальным склерозом, который не определили при биопсии из-за неправильного забора материала.
Полную ремиссию наблюдают у более 80% пациентов, пролеченных кортикостероидами, и лечение обычно продолжают в течение 1-2 лет. Однако, не менее половины рецидивов требуют лечения по такой же или другой схеме.
Цитотоксические лекарственные средства для перорального приема
У нереспондентов, которые принимали кортикостероиды ( < 5% детей и > 10% взрослых), у которых частые рецидивы, и кортикостероид-зависимых пациентов длительная ремиссия может быть достигнута с помощью орального цитотоксического препарата (обычно циклофосфамида от 2 до 3 мг/кг 1 раз в день в течение 12 недель или хлорамбуцил 0,15 мг/кг 1 раз в день в течение 8 недель). (см. the Cochrane abstract review Non-corticosteroid treatment for nephrotic syndrome in children). Тем не менее, эти препараты могут подавлять функцию гонад (самая серьезная в препубертатном периоде), вызывают геморрагический цистит, имеют мутагенный потенциал, и подавляют костный мозг и функцию лимфоцитов. Дозировку препарата следует контролировать с помощью частых клинических анализов крови, а геморрагический цистит выявлять в анализе мочи. Взрослые, особенно пожилые или с артериальной гипертензией, более склонны к побочным действиям этих цитотоксических препаратов. Другая альтернатива - это циклоспорин перорально 3 мг/кг 2 раза в день, доза должна быть приспособлена для получения концентрации в цельной крови 50-150 мкг/л (40-125 нмоль/л). Пациенты, отвечающие на терапию циклоспорином, часто претерпевают рецидив при прекращении приема препарата.
Другие виды лечения
Большинство пациентов, устойчивых к этим видам терапии, отвечает на альтернативные методы лечения, включая ингибиторы ангиотензин-превращающего фермента (АПФ), азатиоприн и мофетила фикофенолат; у менее 5% пациентов заболевание прогрессирует до почечной недостаточности.
Ключевые моменты
Гломерулонефрит с минимальными изменениями является причиной большинства случаев нефротического синдрома у детей и, как правило, носит идиопатический характер.
Следует подозревать болезнь минимальных изменений у детей с внезапно проявившейся протеинурией в нефротическом диапазоне при нормальной функции почек, а также при анализе осадка мочи не нефритического характера.
Необходимо подтвердить диагноз с помощью биопсии почки у взрослых и атипичных случаев заболевания у детей.
Обычно достаточно лечения кортикостероидами.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Болезнь Беста - симптомы и лечение
Что такое болезнь Беста? Причины возникновения, диагностику и методы лечения разберем в статье доктора Рытик Нины Петровны, офтальмолога со стажем в 13 лет.
Над статьей доктора Рытик Нины Петровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Болезнь Беста — это генетическое заболевание глаз, которое вызывает изменения в центральной области сетчатки, так называемой макуле. Первоначально оно протекает бессимптомно, но постепенно приводит к ухудшению остроты зрения и появлению слепого участка в центре поля зрения.
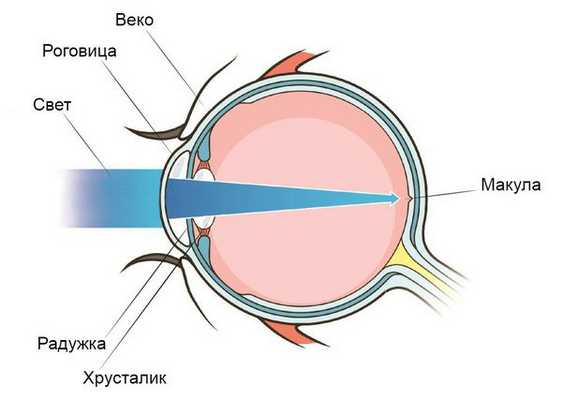
Возникновение и развитие болезни связано с дефектами в наследственном аппарате — мутациями гена VMD2 [3] . Область, в которой локализуется ген VMD2, известна врачам как область скопления генов, отвечающих за развитие большой группы гетерогенных генетических заболеваний с поражением глаз в комплексе с умственной отсталостью , ожирением , тугоухостью и другими патологиями органов и систем [11] .
Болезнь Беста наследуется по аутосомно-доминантному пути, т. е. вероятность возникновения заболевания не зависит от пола и тяжести течения болезни у родителей.
У данного заболевания высокая пенетрантность, т. е. болезнь развивается у большинства людей с патологическим геном. Несмотря на это, выраженность клинических проявлений у родственников варьирует от минимальных изменений и сохранения 100 % зрения до выраженного снижения остроты зрения (вплоть до 0,02) и инвалидизации. Проследить семейную отягощённость удаётся не всегда, даже при комплексном обследовании [2] .
Заболевание считается редким. Его распространённость составляет около 1 случая на 10000 населения [1] . Впервые упоминание о нём в литературе появилось в 1905 году: офтальмолог Ф. Бест описал его как двустороннюю макулодистрофию, которую он наблюдал у восьми членов одной семьи.
Факторов, способствующих манифестации заболевания при наличии наследственной предрасположенности, не выявлено.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы болезни Беста
На начальных стадиях заболевание никак не проявляется, поэтому обычно оно диагностируется случайно при медицинском осмотре. Симптомы появляются только на более поздних стадиях патологии.
Манифестирует болезнь в возрасте 5-15 лет. Ребёнок жалуется на прогрессирующее, обязательно двустороннее снижение чёткости зрения: он начинает испытывать затруднение при чтении мелкого шрифта вблизи. Также его беспокоят "вспышки", "мерцание огоньков" и ухудшение цветовосприятия (в основном наблюдается цветослабость на жёлто-зелёный, зелёный, сине-зелёный цвета).
Изменения в большинстве случаев несимметричные, но возникают на обоих глазах. Острота зрения в зависимости от стадии болезни значительно колеблется: от 0,01 до 1,0.
Важно отличать болезнь Беста от вителлиформной макулодистрофии взрослых. Это заболевание также является генетическим, но возникает и прогрессирует оно в зрелом возрасте (около 40-50 лет). Для него характерно двустороннее, как правило, симметричное субретинальное поражение в центральной области сетчатки. Изменения так же, как при болезни Беста, имеют локальную, очерченную, круглую форму, желтоватый оттенок, но их размер всегда значительно меньше и они не прогрессируют [10] .
Патогенез болезни Беста
Патофизиологический механизм болезни Беста на данный момент неизвестен. Предполагается, что при данной патологии происходит аномальное накопление вещества, подобного липофусцину — жёлто-коричневому пигменту гликопротеиновой природы, который встречается во всех тканях и органах человека. Это вещество скапливается между пигментным эпителием сетчатки и слоем светочувствительных клеток — фоторецепторов. В результате нарушается мембрана Бруха (самый внутренний слой сосудистой оболочки глаза) и разрушаются нервные элементы сетчатки.
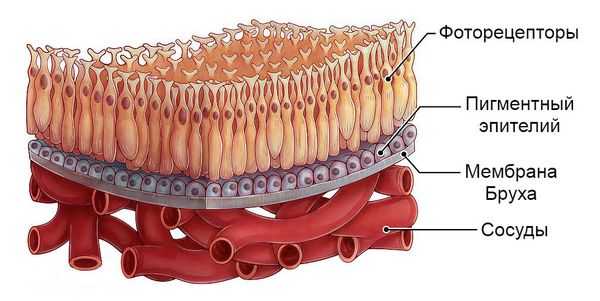
Большинство наружных сегментов фоторецепторов разрушается полностью, при этом во внутренних сегментах светочувствительных клеток скапливаются продукты кислых мукополисахаридов, вызывая изменения в сетчатке. Слой пигментного эпителия отделяется от слоя фоторецепторов, из-за чего формируется немного проминирующее (выбухающее) жёлтоватое образование, т.е. киста, которая локализуется в центральной или околоцентральной области сетчатки [3] .
Классификация и стадии развития болезни Беста
Заболевание относится к наследственным ретинальным дистрофиям. В Международной классификации болезней (МКБ-10) ему присвоен код H35.5.
Болезнь Беста всегда протекает с прохождением четырёх определённых стадий, которые завершаются образованием рубца в центральной области сетчатки [1] .
- Первая стадия — превителлиформная. Она предшествует формированию кисты. В макуле наблюдаются изменения пигментации в виде множества маленьких желтоватых очажков. Во время электроокулографии фиксируются аномальные изменения.
- Вторая стадия — вителлиформная. Формируется классическая вителлиформная киста, которая при осмотре очень похожа на "яичный желток" довольно крупных размеров (до 1 диаметра диска зрительного нерва). Изменения асимметричны и практически всегда поражают оба глаза. Офтальмоскопические изменения не соответствуют высокой остроте зрения: даже у пожилых пациентов этот показатель находится в пределах от 0,4 до 0,9.
- Третья стадия — разрыв кисты и её резорбция, т. е. рассасывание. Острота зрения снижается, пациентам становится трудно читать мелкий текст.
- Четвёртая стадия — формирование фиброглиального рубца на месте кисты. Также на этой стадии возможно развитие субретинальной неоваскуляризации — образование новых сосудов под сетчаткой [3] . Острота зрение падает ещё сильнее, в поле зрения появляются слепые зоны.
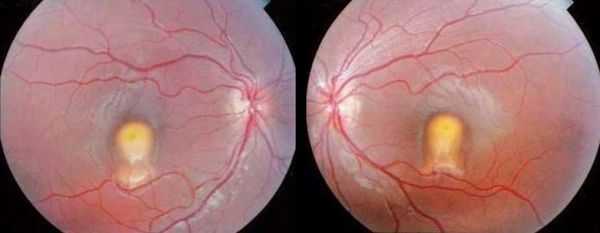
Осложнения болезни Беста
На поздних стадиях заболевания начинают развиваться осложнения. Самые частые — это образование сквозных разрывов в макулярной области, потеря центральной остроты зрения и формирование "пятна перед глазами". Возникновение отслойки сетчатки встречается реже.

Одним из частых осложнений так же является субретинальная неоваскуляризация — образование новых сосудов в зоне глазного дна [11] . Она является одной из причин инвалидизации при болезни Беста, так как приводит к стойкому снижению центральной остроты зрения. При этом полная слепота не развивается.
Диагностика болезни Беста
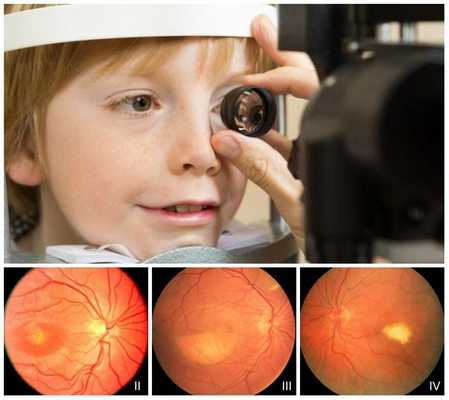
Для постановки диагноза "болезнь Беста" необходим ряд обследований. В первую очередь нужно зарегистрировать специфическую офтальмоскопическую картину глазного дна, характерную для каждой стадии заболевания. Затем, чтобы оценить состояние сетчатки, следует выполнить электроокулографию (ЭОГ) и электроретинографию (ЭРГ). В последние годы, с развитием генетики, стало обязательным молекулярное генетическое исследование пациента. Вместе с этим производят офтальмологическое и генетическое обследование других членов семьи.
Офтальмоскопия позволяет обнаружить первые признаки болезни Беста на первом и втором десятилетии жизни. На первых стадиях в макуле появляются желтоватые очажки, затем формируются виттелиформные кисты, при разрывах которых глазное дно напоминает картину "яичницы-глазуньи". На последних стадиях в макуле формируется атрофические глиальные рубцы, а при осложнениях — неоваскулярная мембрана и сквозные макулярные отверстия.
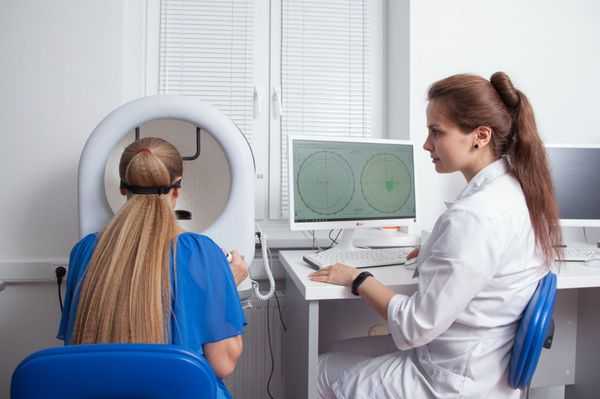
Электроокулография является очень важным методом диагностики при болезни Беста. С её помощью можно выявить такой специфический признак заболевания, как снижение коэффициента Ардена — соотношения биопотенциала глаз в темноте и при свете. Данный показатель будет снижен даже на нулевой стадии болезни при нормальной офтальмоскопической картине. В случае вителлиформной макулодистрофии взрослых электроокулография изменений не выявит.

Флюоресцентная ангиография позволяет диагностировать болезнь Беста даже на доклиническом этапе. Изменения сетчатки зависят от стадии заболевания. На начальном этапе болезни видны мелкие окончатые дефекты и участки с локальной гиперфлюоресценцией (свечением). При наличии сформированной кисты флюоресценция полностью отсутствует. На третьей стадии болезни гиперфлюоресценция наблюдается только в верхней части кисты, а в нижней её части отсутствует [8] .
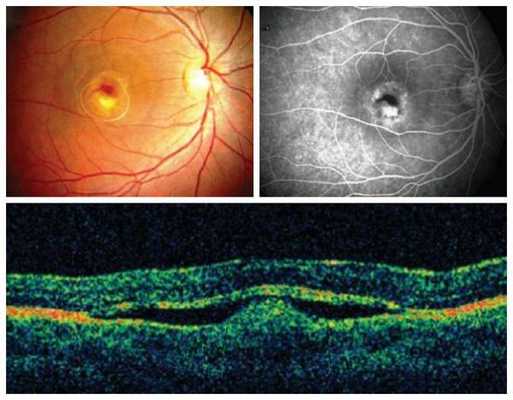
При электроретинографии (общей и ритмичной) наблюдается снижение амплитуды "а" волн, увеличение латентных периодов "а" и "b" волн, а также угнетение ответа от центральных отделов сетчатки [9] .
Статическая компьютерная периметрия выявляет снижение чувствительности макулярной области, а при разорвавшейся вителлиформной кисте — центральную абсолютную скотому (слепое пятно) . При этом границы поля зрения по периферии остаются в норме.
Дифференциальная диагностика
Болезнь Беста необходимо отличить от других заболеваний:
- болезни Коатса и других макулярных дистрофий пигментного эпителия сетчатки (Х-образной макулярной дистрофии);
- острого хориоретинита (в первую очередь токсоплазмозной этиологии);
- болезни Штаргардта;
- вителлиформной дистрофии взрослых;
- отслойки пигментного эпителия сетчатки [11] .
Болезнь Коатса — врождённая ненаследственная патология, при котором наблюдаются аномалии сосудов сетчатки глаза. Она может стать причиной частичной или полной слепоты. На первых стадиях болезнь Коатса так же, как и болезнь Беста, течёт без симптомов и в большинстве случаев диагностируется у детей 2-8 лет при случайных профилактических осмотрах, проводимых в детских садах или школах. В отличие от болезни Беста, при болезни Коатса, как правило, в патологический процесс вовлекается только один глаз. Лишь у 5-8 % больных наблюдаются двусторонние изменения. Мальчики заболевают им в 3 раза чаще, чем девочки [9] .
У большинства пациентов с заболеванием Коатса во время первого офтальмологического обследования в центральной области сетчатки выявляют возвышающийся округлый очаг твёрдого желтоватого экссудата, очень похожий на вителлиформные изменения, встречающиеся при болезни Беста. Данные экссудаты в центральной области сетчатки очень часто сочетаются с кровоизлияниями за сетчаткой и образованием новых сосудов, что тоже характерно для болезни Беста.
Диагностика болезни Коатса основывается на результатах осмотра глазного дна (офтальмоскопии), а также центральной области и периферии глазного дна с применением или налобного офтальмоскопа с линзами 20-30 дптр, или офтальмобиомикроскопии с трёхзеркальной линзой Гольдмана. У большинства пациентов с болезнью Коатса во время данного обследования обязательно выявляются аномалии сосудов: микро- и макроаневризмы, телеангиэктазии ( сосудистые звёздочки ), артериовенозные шунты. Чаще всего эти аномалии располагаются в височной половине сетчатки.
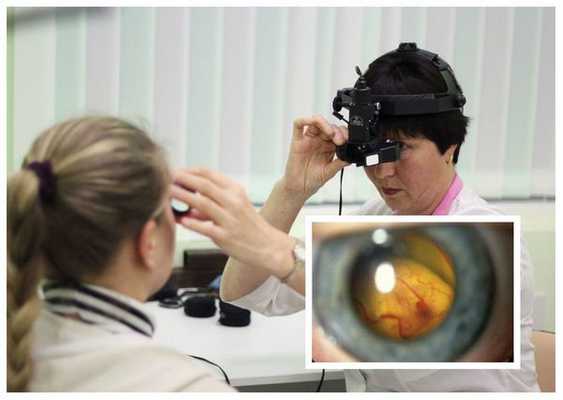
Изменения при электроокулографии в начальной и развитой стадиях заболевания, как и при болезни Беста, не выявляются. Но острота зрения у больных с ретинитом Коатса низкая: при наличии изменений в центральной области сетчатки она не превышает 0,4, а при болезни Беста длительно сохраняются в норме [7] .
Острый токсоплазмозный хориоретинит — это паразитарное заболевание глаз, для которого характерно в основном скрытое или хроническое течение. Как правило, оно поражает только один глаз, в отличие от болезни Беста. При неадекватном и несвоевременном лечении может привести к стабильному снижению остроты зрения.
Трудности при диагностике обычно возникают при обследовании больных с приобретённым токсоплазмозом и экссудативно-геморрагическими изменениями в центральной области сетчатки, которые похожи на вителлиформные очажки с субретинальными геморрагиями, как при болезни Беста. Хориоретинальные поражения при токсоплазмозе сочетаются с выпотом в стекловидное тело различной степени тяжести, а порой и изменениями переднего отрезка глаза.
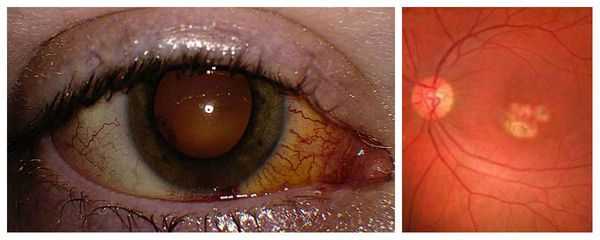
Пациенты, как правило, предъявляют жалобы на внезапное, резкое и значительное снижение центральной остроты зрения, которая в ряде случаев изменяется до 0,01-0,2. При статической периметрии регистрируется повышенный порог яркостной чувствительности, относительная или абсолютная центральная скотома. Электроокулография, как правило, не изменена.
Для подтверждения и верификации токсоплазмоза необходимо выполнить иммунологические исследования, определить антитела к токсоплазме или выделить её методом ПЦР (полимеразной цепной реакции). В 95 % случаев токсосплазмозный хориоретинит хорошо поддаётся лечению, острота зрения может восстановиться до исходных значений [12] .
Болезнь Штаргардта иначе называют ювенильной макулярной дегенерацией. Впервые это заболевание описал немецкий врач-офтальмолог К. Штаргардт как врождённое поражение макулы, которое передавалось по наследству в одной семье.
Как правило, болезнь Штаргардта начинается в молодом (ювенильном) возрасте и сопровождается поражением центральной области сетчатки, как и болезнь Беста. Её отличает быстрое прогрессирование, вплоть до инвалидизации. Медленное ухудшение зрения встречается крайне редко (при доминантном типе наследования) [9] .
Данное заболевание всегда двустороннее и симметричное. Его характерными признаками являются различные процессы: атрофия хориоидеи (сосудистой оболочки глаза), "бычий глаз", очаг дистрофии цвета "битой (кованой) бронзы" и др. При далеко зашедшем процессе фовеолярный рефлекс (центральная ямка макулы) отсутствует. В макулярной области на уровне пигментного эпителия сетчатки наблюдаются скопления коричневатого, тёмного пигмента, который окружён участками гипер- и депигментации. Эти скопления отличаются от вителлиформных жёлтых очагов при болезни Беста.
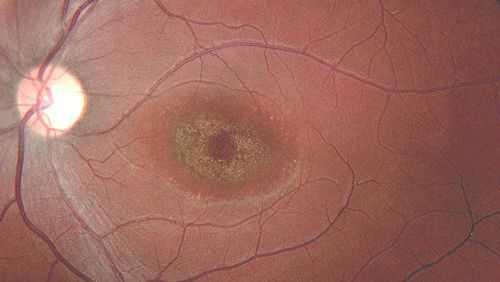
Вителлиформная макулодистрофия взрослых , в отличие от болезни Беста, развивается у людей 40-50 лет. Для неё характерно двустороннее симметричное поражение макулы с локальными изменениями. Очаги патологии, как правило, имеют округлую форму и жёлтый оттенок. Их отличает отсутствие прогрессирования и небольшие размеры: диаметр поражений достигает 0,3-0,5 размера диска зрительного нерва. При этом нарушения зрительных функций минимальны.
Вещества, подобные липофусцину, скапливаются диффузно в различных местах: в пигментном эпителии, внутренней части фоторецепторов, мюллеровских клетках и даже в стекловидном теле.
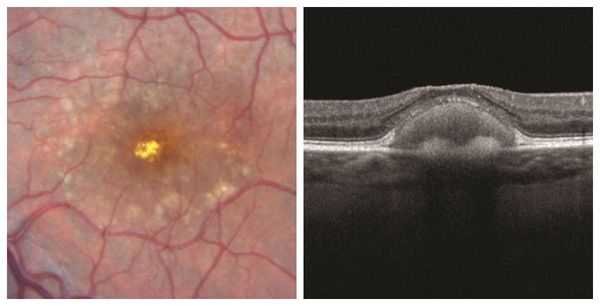
Отслойка пигментного эпителия сетчатки возникает при нарушении нормального соединения пигментного эпителия сетчатки с мембраной Бруха. К причинным факторам относятся сосудистые, воспалительные и дистрофические процессы.
Длительное время болезнь может существовать без какой-либо динамики, спонтанно исчезать, а вследствие инвазии сосудов из хориоидеи трансформироваться в геморрагическую отслойку. Сложности в диагностике могут возникнуть при атипичной клинической картине заболевания и экссудативных изменениях в центральной области сетчатки.
Обычно болезнь поражает только один глаз. Общая электроретинография не меняется, локальная — может быть снижена. Электроокулография, как правило, в норме. При флюоресцентной ангиографии на последней стадии отслойки регистрируется гиперфлюоресценция.
Лечение болезни Беста
Лечения болезни Беста на сегодняшний момент не существует. Можно лишь затормозить формирование кист, предотвратить их разрывы и наступление осложнений, которые сильно влияют на остроту зрения пациента. С этой целью применяется антиоксидантная и вазоактивная терапия, а также витамины С, Е и А. Вазоактивные препараты улучшают кровоснабжение сетчатки, а витамины являются мощными антиоксидантами [5] .
Вероятно, что в будущем при лечении болезни Беста будет применяться генная инженерия или терапия, направленная на нормализацию тока жидкости через пигментный эпителий сетчатки, торможение накопления липофусцина и воздействия на другие этапы патогенеза [13] .
Прогноз. Профилактика
Прогноз для зрения относительно благоприятный. Так как заболевание протекает медленно, острота зрения снижается постепенно, при этом страдает только центральное зрение. Благодаря сохранению периферического зрения человек хорошо адаптируется, часто сохраняя работоспособность и возможность чтения.
При обнаружении заболевания врач должен проинформировать больного о вариабельности течения заболевания, рисках его возникновения у будущих детей и функциональных прогнозах. Желательно проконсультироваться с врачом-генетиком.
Все члены семьи пациента обязательно должны пройти обследование у офтальмолога. Сам пациент должен регулярно наблюдаться у своего лечащего врача [14] .Чтобы дети и подростки не отставали в развитии, необходимо вовремя применять технические средства реабилитации слабовидящих [1] .
Научная электронная библиотека

Гломерулонефрит (ГН) гетерогенная группа иммуно-воспалительных заболеваний преимущественно клубочкового аппарата почек с различной клинико - морфологической картиной, течением и исходами.
В настоящее время в развитии и прогрессировании хронического гломерулонефрита придают значение трем основным механизмам: иммунным, гемодинамическим и метаболическим. Однако в развитии гломерулонефрита наибольшее значение имеет первый из этих факторов. В соответствии с данными литературы, механизм развития гломерулярной патологии, прежде всего, должен рассматриваться как следствие генетической неполноценности Т-клеточного звена иммунитета, что в конечном итоге приводит к нарушению процессов репарации отдельных частей нефрона с дальнейшим изменением их антигенной структуры и образованием иммунных комплексов. Последние локализуются на пораженных участках базальной мембраны с последующим их фагоцитозом подоцитами, нейтрофилами, мезангиальными клетками и макрофагами. При разрушении некоторых из этих клеток выделяются лизосомальные ферменты, в результате чего базальная мембрана повреждается еще больше. К этому предрасполагает низкое содержание Т-лимфоцитов,
что делает процесс необратимым. В литературе рассматриваются несколько вариантов гломерулонефрита:
1. Нефритический - проявляется гематурией, протеинурией, гипертензией, олигурией, цилиндрурией, лейкоцитурией, гиповолемией, гипокомплементемией.
2. Нефротический - высокая протеинурия, отеки, гипопротеинемия, возможно артериальная гипертензия, эритроцитурия, азотемия.
3. Смешанный - выраженный нефротический синдром, значительная гематурия, гипертензия.
Современные данные о гломерулонефрите у детей можно получить из клинических рекомендаций:
Клинические рекомендации по оказанию медицинской помощи детям с нефротическим синдромом, 2015. Союз педиатров России.
Клинические рекомендации «Диагностика и лечение болезни минимальных изменений у детей», 2014.
Ассоциация нефрологов Творческое объединение детских нефрологов Научное общество нефрологов России.
Определение
Нефротический синдром (НС) - клинико-лабораторный симптомокомплекс, характеризующийся протеинурией (> 50 мг/кг/сут или > 40 мг/м2/час, т. е. 2,5 г/сут и более), гипоальбуминемией (< 25 г/л), диспротеинемией, гиперлипидемией, отеками, в том числе полостными.
Коды по МКБ-10
N04 - Нефротический синдром.
Классификация
Выделяют первичный и вторичный Нефротический синдром (НС).
- Первичный НС развивается при заболеваниях собственно клубочков почек.
- Вторичный НС может быть обусловлен обширной группой заболеваний, вызывающих формирование специфической нефропатии.
Основные формы первичного НС:
- Врождённый НС (дебют до 3-месячного возраста).
- Врождённый НС финского типа.
- Инфантильный НС (дебют в возрасте от 3 месяцев до 1 года).
- Идиопатический НС.
Основные причины вторичного НС:
- Наследственные заболевания (поликистоз почек, синдром Альпорта, спондилоэпифизарная дисплазия, болезнь Фабри, синдром Марфана и пр.).
- Коллагенозы (системная красная волчанка, системная склеродермия, дерматомиозит, ревматоидный артрит, ревматизм).
- Системные васкулиты (геморрагический васкулит, узелковый полиартериит, гранулематоз Вегенера).
- Гемолитико-уремический синдром.
- Рефлюкс-нефропатия.
- Амилоидоз почек.
- Сахарный диабет.
- Болезни крови (лимфогранулематоз, смешанная криоглобулинемия, миеломная болезнь, серповидно-клеточная анемия, талассемия).
- Тромбозы вен и артерий почек, аорты или нижней полой вены.
- Опухоли различной локализации.
- Лекарственное поражение почек (препараты висмута, золота, противоэпилептические препараты и др.), введение вакцин.
- Болезни вирусной этиологии (гепатит B и C, цитомегаловирусная инфекция, ВИЧ-инфекция).
- Болезни бактериальной этиологии (септический эндокардит; пневмония, абсцессы, бронхоэктазы, остеомиелит; туберкулёз, сифилис).
В зависимости от ответа на стандартный курс терапии преднизолоном НС принято делить на:
- Стероидчувствительный НС.
- Стероидчувствительный стероидзависимый НС.
- Стероидрезистентный НС.
Клиническая картина
Первым клиническим симптомом, заметным для больного и окружающих, являются отеки. Они могут развиться постепенно или же стремительно, достигнув степени анасарки. Периферические отеки выявляются в области век, лица, поясничной области и половых органов, могут распространяться на всю подкожную клетчатку, растягивая кожу до образования striae distensae. В это время у больных могут образовываться транссудаты в серозные полости: одно- или двусторонний гидроторакс, асцит, гидроперикард; возможно развитие отека легких.
При обследовании пациента обязательным является измерение артериального давления, которое может быть повышено у детей с активной стадией нефротического синдрома.
Диагностика
Диагноз устанавливается на основании наличия триады симптомов:
- протеинурия более 50 мг/кг/сут;
- гипоальбуминемия менее 25 г/л;
В норме у здоровых людей может обнаруживаться протеинурия, но не более 150 мг/сут.
Лабораторные исследования
Суточная экскреция белка с мочой > 50 мг/кг/сут.
Гематурия не характерна для нефротического синдрома.
Гипопротеинемия: общий белок крови снижается до 40-30 г/л.
Гиперлипидемия: наиболее характерно повышение содержания в сыворотке крови холестерина, триглицеридов. Может быть повышен креатинин, что является следствием гиповолемии при нефротическом синдроме. Снижение уровня электролитов (гипонатриемия, гипокальциемия).
Коагулограмма: повышение уровня фибриногена, снижение уровня антитромбина III в сыворотке крови, тромбоцитоз.
Общий анализ крови: высокая СОЭ является признаком активности нефротического синдрома и гипопротеинемии. Лейкоцитоз может быть следствием как приема кортикостероидных препаратов, так и проявлением бактериальной инфекции, которая часто осложняет течение нефротического синдрома.
К дополнительным лабораторным исследованиям, используемым для уточнения генеза нефротического синдрома, относят:
- вирусологические исследования: маркеры вирусов гепатита В, С, дельта;
- иммунологическое исследование крови: анти-ДНК, АНФ, С3-фракция комплемента, АСЛ-О, криоглобулины;
- генетическое исследование при стероидрезистентном нефротическом синдроме для определения мутации генов нефрина (NPHS1) и подоцина (NPHS).
Обязательные инструментальные исследования
1. Измерение АД, в том числе суточное мониторирование АД.
2. Эхо-КГ: оценка морфометрических параметров сердца и крупных сосудов при отеках, артериальной гипертензии, выявление гидроперикарда.
3. ЭКГ: выявление признаков возможных электролитных нарушений.
4. УЗИ почек (с допплерографией внутрипочечных сосудов).
Показания к биопсии почки при нефротическом синдроме:
- стероидрезистентность нефротического синдрома (первичная и вторичная);
- НС у детей младше 1 года и старше 12 лет;
- через 2-2,5 года после лечения циклоспорином А.
Лечение
Цель терапии: Снижение активности или достижение ремиссии нефротического синдрома.
Медикаментозное лечение
Кортикостероиды (КС): Стандартный курс преднизолонотерапии - пероральный прием Преднизолона 2 мг/кг/день (60 мг/м2), максимальная доза - 60 мг/сутки, непрерывно в течение 4-6 недель. Далее проводится терапия КС в альтернирующем режиме, т. е. через день в дозе 2/3 от лечебной (1,5 мг/кг/сутки или 40 мг/м2, но не более 40 мг/сутки по преднизолону). Длительность альтернирующего режима приема КС составляет 4-6 недель. После завершения этого курса проводят постепенное снижение дозы по 10 мг/м2 в 7-10 дней до полной отмены. Общая длительность терапии КС должна составлять 4-5 месяцев.
Симптоматическая терапия
Диуретические препараты широко используются для лечения больных с отеками. Наиболее часто с этой целью применяются петлевые диуретики: Фуросемид в возрастной дозировке. Многим больным с активным нефротическим синдромом, гипоальбуминемией и рефрактерными отеками для получения адекватного диуреза, помимо петлевых диуретиков, необходимо внутривенное введение 20 % раствора альбумина под контролем уровня АД, ЧСС и Эхо - кардиографических показателей сосудистого объема.
Гипотензивная и нефропротекторная терапия. Блокада ренин-ангиотензин-альдостероновой системы ингибитрами ангиотензин превращающего фермента (иАПФ): Фозиноприл, Эналаприл индивидуальный подбор дозы, в среднем: 0,1-0,3 мг/кг по Фозиноприлу и блокаторами ангиотензиновых рецепторов (БАР) оказывают гипотензивный, антипротеинурический и антисклеротический эффект.
Также могут применяться блокаторы медленных кальциевых каналов: Амлодипин.
Показания к госпитализации
Все дети в активную стадию нефротического синдрома должны быть госпитализированы в специализированное отделение. Дети в стадии ремиссии могут наблюдаться в амбулаторных условиях с регулярным (1-2 раза в год) стационарным специализированным обследованием в условиях круглосуточного или дневного пребывания.
Длительность пребывания в стационаре составляет в среднем 14-21 день при дебюте и рецидивах нефротического синдрома, также показана плановая госпитализация с целью контрольного обследования и коррекции терапии - 1 раз в 6 месяцев. Амбулаторно проводится контроль лабораторных показателей: уровень протеинурии, клиническим и биохимическим анализом крови, коагулограммой (частота обследования определяется индивидуально, в зависимости от состояния ребенка).
Болезнь минимальных изменений (БМИ) - это непролиферативная гломерулопатия, не имеющая каких-либо морфологических критериев при световой микроскопии, обусловленная повреждением (иммунным или неиммунным) подоцитов (подоцитопатия), которое диагностируется исключительно при ультраструктурном анализе в виде диффузного слияния ножковых отростков подоцитов. Повреждение подоцита определяет формирование в клинике заболевания нефротического синдрома (НС).
БМИ составляет 76,6 % всех морфологических вариантов первичного гломерулонефрита (ГН) у детей.
- Наибольшая встречаемость у детей раннего возраста.
- БМИ чаще отмечается у мальчиков в соотношении 2:1.
- Возможны семейные формы, обусловленные мутациями генов структурных белков подоцита.
Первичная (идиопатическая) БМИ
Основой развития идиопатического нефротического синдрома у детей является дисфункция Т-клеточного звена иммунной системы или генетические мутации. Однако БМИ может быть ассоциирована с множеством других патологических состояний таких, как аллергия, онкопатология, лекарственные воздействия.
Клиническим синдромом БМИ является внезапно развившийся НС. Отягощенный аллергологический анамнез и аллергические проявления у детей с БМИ наблюдаются в 30-70 % случаев в отличие от других форм гломерулонефрита. Триггерными факторами могут быть ОРВИ, детские инфекции, атопические реакции.
- Артериальная гипертензия наблюдается крайне редко и характеризуется кратковременностью. Повышение артериального давления при БМИ связано с компенсаторным механизмом на выраженную гиповолемию. При резкой гиповолемии возможно развитие нефротического криза с болями в животе, кожной эритемой и сердечно-сосудистым шоком с циркуляторной недостаточностью.
- Течение БМИ делится на острое с исходом в стойкую ремиссию (20-30 %), рецидивирующее и часто рецидивирующее течение. По отношению к стероидной терапии выделяют следующие формы: стероидчувствительная, стероидзависимая и стероидрезистентная.
Прогноз
В целом, при БМИ при наличии чувствительности к кортикостероидам, отдаленный прогноз, как правило, благоприятный, и у большинства детей пациентов развивается стойкая ремиссия. Прогноз в отношении почечной функции также благоприятный.
Факторами неблагоприятного прогноза являются наличие генетически обусловленного БМИ.
Читайте также:
- Скрининг риска синдрома Дауна. Снижение количества инвазивных диагностических вмешательств
- Выдергивание волос (трихотилломания) ребенком
- Кровотечение желудочно-кишечное: причины, симптомы и лечение
- Привлечение больных шизофренией к труду. Выдача власти больным шизофренией
- Симптомы и клиника болезней средней черепной ямки