Буллезный эпидермолиз (БЭ) у ребенка
Добавил пользователь Morpheus Обновлено: 21.01.2026
Фонд «Дети-Бабочки» совместно с Национальным медицинским исследовательским центром акушерства, гинекологии и перинатологии имени академика В.И. Кулакова запустили совместный проект по оказанию комплексной специализированной помощи детям с буллезным эпидермолизом и их семьям
На сегодняшний день буллезный эпидермолиз (БЭ) - неизлечимое заболевание с хроническим течением. Возможности современной медицины, по крайней мере пока, сводятся к симптоматической терапии, правильному уходу и поиску новых протоколов лечения, которые позволили бы лучше справляться с проявлениями БЭ, предотвращать (или хотя бы отодвинуть во времени) тяжелые осложнения и повысить качество жизни пациентов.
Уже почти 11 лет фонд «Дети-Бабочки» оказывает комплексную помощь пациентам с буллезным эпидермолизом и их семьям: обеспечивает медицинскую, психологическую и юридическую поддержку, медикаменты, патронаж, обучение врачей. Совместно с ФГБУ «Национальный Медицинский Исследовательский Центр акушерства, гинекологии и перинатологии имени академика В.И. Кулакова» Министерства здравоохранения Российской Федерации (ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ) фондом был инициирован специальный проект, который позволит детям с БЭ по всей России получить высококачественную медицинскую помощь.
Центр имеет большие диагностические мощности, активно использует инновационные технологии лечения, в частности, клеточную терапию. Фонд «Дети-Бабочки», в свою очередь, обладает организационными компетенциями и наработками в области ухода за детьми с буллезным эпидермолизом. Объединение усилий дает группе врачей и исследователей шанс на разработку новых протоколов лечения.
«Мы можем «подхватить» маленького пациента в самом начале его жизни и дать задел на будущее, ресурс для организма, чтобы как можно дольше избегать осложнений, таких как длительно незаживающие раны, срастание пальцев, контрактуры и тому подобное» - подчеркивает Юлия Юрьевна Коталевская, врач-генетик фонда.
В проекте участвуют дети с самой тяжелой дистрофической формой БЭ. Первый этап программы - проведение телемедицинского консилиума регионального врача и специалистов ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ. В процессе телеконсультации оценивается состояние новорожденного, согласовывается тактика медицинской помощи, определяется тактика ухода за кожей, обсуждается необходимость перевода ребенка в Москву. После дифференциальной диагностики ребенок госпитализируется в ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ, где разрабатывается индивидуальный план лечения.
Фонд финансирует собственно научную составляющую проекта, затраты на генетическую диагностику и медикаменты на весь период госпитализации. Пребывание в стационаре ребенка с мамой, необходимые исследования, лечение других заболеваний и уход производятся за счет средств ОМС. Транспортные расходы покрываются за счет региональных департаментов здравоохранения и средств благотворительного фонда «Дети-бабочки».
Программа пребывания в стационаре объединяет в себе две главные составляющие:
- правильный уход с первых дней жизни, который поможет изначально избежать серьезных (и, к сожалению, частых) ошибок в ведении пациентов с БЭ;
- терапию с применением клеточных технологий, направленную на стимуляцию процессов регенерации и поддержание стабильности кожного покрова.
Специалисты фонда курируют лечение на всех его стадиях и находятся в постоянном контакте с врачами в отделении и мамой пациента.
После поступления ребенку назначается комплексное обследование и консультация узкопрофильных специалистов. К моменту перевода пациента в отделение фонд организует доставку специальных перевязочных материалов. Дополнительно под контролем медиков маму обучают основам ухода за ребенком.
Изначально проект был рассчитан на три года, но пандемия «скорректировала» его продолжительность. Тем не менее, в 2022 году планируется публикация промежуточных данных, которые в первом приближении позволят сделать выводы об эффективности протоколов клеточной терапии.
Исследования по поиску эффективных способов борьбы с патологией кожи проводятся во всем мире. Совместный проект Фонда и ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ по созданию мультидисциплинарного подхода в сочетании с клеточными технологиями - один из шагов в этом направлении.
Буллезный эпидермолиз
Буллезный эпидермолиз - группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10
Общие сведения
Буллезный эпидермолиз - это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса - десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии - пограничного буллезного эпидермолиза - являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз - имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз - делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз - имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи - эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе - киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно - развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты - механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств - типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность - как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Люди-бабочки: что такое буллезный эпидермолиз и как наука ищет способы его лечить
Как устроена наша кожа и из-за чего возникает болезнь, при которой кожа становится очень хрупкой и может повредиться от любого прикосновения? Как живут люди, которых называют «бабочками», и чем медицина может им помочь?
Вместе с информационно-просветительским гуманитарным проектом «12 месяцев» мы продолжаем серию материалов о редких (орфанных) генетических заболеваниях и жизни людей с ними.
Читайте в январе рассказ о буллезном эпидермолизе, который встречается у одного из 100 тысяч человек, а также историю юриста Игоря Чувствинова.
Что важно знать о буллезном эпидермолизе
Буллезный эпидермолиз (БЭ) — это группа генетических заболеваний, при которых даже незначительное механическое воздействие на кожу и слизистые оболочки приводит к образованию пузырей. Когда эти пузыри разрываются, на их месте остаются болезненные раны, которые быстро инфицируются и с трудом поддаются лечению.
Болезнь сильно снижает качество жизни людей и требует сложного и дорогостоящего ухода. Нередко проявления заболевания выражены настолько, что пациенты полностью зависят от своих близких.

Источник: сайт фонда «Дети-бабочки»
Точно посчитать частоту встречаемости буллезного эпидермолиза сложно. По данным Национального института здоровья США, заболеваемость БЭ составляет примерно 1 случай на 100 тысяч человек в общей популяции и почти 2 случая на 100 тысяч новорожденных. В России, по оценкам фонда «Дети-бабочки», который помогает детям с буллезным эпидермолизом, с этим заболеванием проживает около 2000-2500 человек.
Почему возникает и как проявляется буллезный эпидермолиз?
Кожа человека состоит из нескольких слоев клеток — кератиноцитов. Они соединяются и удерживаются вместе с помощью специальных белков, основные из которых — это кератины, коллаген, ламинин и киндлин. Мутации в генах, необходимых для их синтеза, приводят к нарушению функции этих белков. Это приводит к тому, что даже при небольшом натяжении или давлении на кожу ее слои отходят друг от друга и в этом месте образуются пузыри.
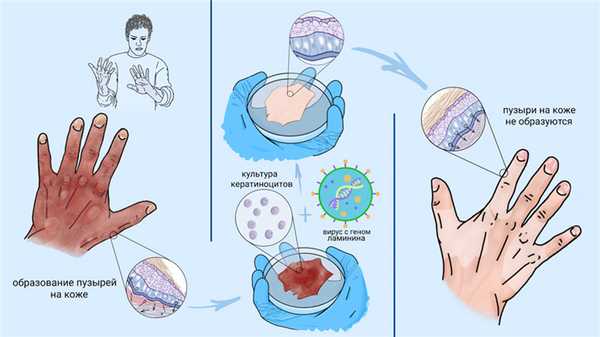
Основные проявления БЭ связаны с кожей, однако болезнь затрагивает весь организм. Так, наиболее часто поражаются слизистые оболочки желудочно-кишечного тракта. В ротовой полости со временем образуются эрозии и пузыри, а в пищеводе — сужения, значительно затрудняющие глотание. Кроме того, нередки случаи нарушения проходимости кишечника, анальных трещин и запоров.
Болезнь поражает и глаза: к ее последствиям относится хроническое воспаление, язвы на роговицах. Способность к самообслуживанию чрезвычайно сильно ограничивает ситуация, когда срастается кожа пальцев.
Еще одна опасность для людей с БЭ — повышенный риск рака кожи, который увеличивается с возрастом.
Можно ли вылечить буллезный эпидермолиз?
Основные способы лечения буллезного эпидермолиза — это ведение образа жизни, уменьшающего вероятность травмирования кожи, и использование специальных дорогостоящих повязок. Однако уже давно у врачей и исследователей появилось желание воздействовать на причину заболевания — неспособность клеток плотно соединяться друг с другом.
Начиная с конца 1980-х годов предпринимались разные попытки лечения, основанные на методах клеточной терапии (то есть терапии стволовыми клетками). Их цель — улучшить течение болезни в тех ситуациях, когда обычное лечение не помогало. Однако в большинстве случаев эффект был выражен слабо и длился очень недолго.
Первым подходом клеточной терапии стала попытка трансплантации пациентам с БЭ кератиноцитов, которые были взяты из непораженных участков кожи. Это дало ограниченный и непродолжительный эффект, который, по-видимому, был обусловлен временным уменьшением воспаления в зоне трансплантации.
Другой подход основывался на внутривенном введении стволовых клеток костного мозга и мезенхимальных стромальных клеток (МСК), способных мигрировать в зоны повреждения. Исследователи предполагали, что эти клетки, трансплантированные от здоровых доноров, синтезируют белки, необходимые для образования нормальной кожи. Однако, как и в предыдущем случае, эффект этого лечения был успешным лишь отчасти.
При использовании стволовых клеток костного мозга наблюдалось временное улучшение течения болезни. Однако несколько пациентов в процессе клинического исследования умерли от осложнений, связанных с подготовкой к трансплантации.
В случае использования мезенхимальных стромальных клеток (МСК) столь драматичных побочных эффектов не было. Но эффект от лечения — ускорение заживления ран и улучшение общего самочувствия — по-прежнему длился не более полугода.
Из-за сложностей при получении стволовых клеток костного мозга и МСК, ученые обсуждают возможность использования индуцированных плюрипотентных стволовых клеток (ИПСК). ИПСК — это любые клетки, которые в лабораторных условиях приводят в т.н. плюрипотентное состояние (когда клетка становится недифференцированной, какой была в эмбриональном периоде развития). Из него они могут стать почти любой другой клеткой, в том числе стволовой клеткой костного мозга, МСК или сразу фибробластом или кератиноцитом. Однако процесс получения клеток из ИПСК пока технически несовершенен, поэтому исследования таких клеток проводятся пока только в условиях лабораторий.
Есть ли надежда на генно-клеточную терапию?
Может сложиться впечатление, что введение стволовых клеток костного мозга и мезенхимальных стромальных клеток (МСК) в системный кровоток малоперспективно из-за технического несовершенства этого подхода и кратковременности терапевтического эффекта. Тем не менее дальнейшие исследования в этом направлении необходимы, потому что на сегодняшний день это единственный подход, потенциально позволяющий действовать на внекожные проявления болезни.

О первом успехе генно-клеточной терапии буллезного эпидермолиза стали говорить в 2006 году, когда группа итальянских исследователей смогла восстановить кожу пациента с пограничным буллезным эпидермолизом. Эта форма заболевания появляется при мутации в гене белка ламинина. Для лечения пациента исследователи выделили из неповрежденных участков его кожи кератиноциты, в которые ввели здоровый ген ламинина. Далее в лабораторных условиях из этих кератиноцитов вырастили тонкие пласты ткани, которые трансплантировали в поврежденные участки кожи. Через полгода кожа в зоне трансплантации не отличалась от нормальной и не требовала повторных вмешательств.
В 2015 году успех этого подхода закрепил опыт по трансплантации практически всего эпидермиса семилетнему мальчику, который к моменту первой трансплантации утратил около 80% кожных покровов.
В результате лечения у пациента сформировалась полностью здоровая и функциональная кожа. Сейчас трансплантированная кожа пациента почти не отличается от здоровой. Он ведет обычную жизнь и даже занимается спортом, хотя до лечения из-за боли практически не мог двигаться, нормально питаться и нуждался в постоянном обезболивании.
- Сегодня эта технология используется в трех препаратах, проходящих клинические исследования — EB-101, LZRSE-Col7A1 и FCX-007. Другой препарат, проходящий клинические исследования — B-VEC. Его преимущество — возможность непосредственного введения в кожу пациентов, что позволяет миновать затратный по времени этап выделения и выращивания клеток пациента.
«Заболевание переориентировало меня на книги»: как жить с буллезным эпидермолизом
26-летний Игорь Чувствинов рассказал, как ему удается учиться, работать, дружить и чувствовать себя счастливым состоявшимся человеком, несмотря на ограничения из-за болезни.

О детстве и постановке диагноза
Дети с буллёзным эпидермолизом нередко имеют слабую кожу уже на момент рождения и получают травмы при родах. Мой случай не был исключением. Но до 3-х лет родителям не понимали, что со мной происходит.
Были две сложности в постановке диагноза: во-первых, врачи первоначально хотели поставить пузырчатку (аутоиммунное заболевание, которое характеризуется появлением пузырей на коже и слизистых оболочках), потому что симптомы этих заболеваний похожи. Во-вторых, врачи принимали проявления болезни за временные явления и ждали, что они пройдут сами, но они не проходили.
Я родился в 1995-м году, и понятно, что всем тогда было не до редких заболеваний. О них не говорили в СМИ, не было профильных фондов. И врачи оказывались в очень сложном положении, видя меня.
На вопрос: «Чем лечиться?» дерматологи отвечали: «Вы столько прожили с этим заболеванием, вы знаете о нем больше, чем я».
О влиянии болезни на жизнь
Я не знаю жизни без этого заболевания, поэтому мне сложно сравнить «до» и «после». Но если смотреть на мою жизнь и жизнь других людей, то, конечно, отличия есть.
Во-первых, я намного чаще наблюдался у врачей.
Во-вторых, точно могу сказать, что мое заболевание повлияло на спектр моих хобби. Я никогда не занимался опасным спортом, не катался на велосипеде, не играл в футбол, не ходил далеко гулять. В детстве это могло быть очень травматичным для моих ног.
Я больше домосед. Заболевание переориентировало меня на книги, фильмы, компьютерные игры и прочее, что не связано с активностью, способной привести к травмам. При этом я довольно общительный человек, поэтому не могу сказать, что болезнь мешала или мешает мне в социализации.
В-третьих, мой диагноз влияет на то, что я ем: я не употребляю продукты, которые могут вызвать аллергическую реакцию. После чего кожа может начать чесаться. И есть риск нарваться на круговорот болячек в природе, когда они уже не заживают, потому что постоянно их расчесываешь. Естественно, что в детстве за этим следили родители, а потом я стал сам видеть связь между употреблением какого-то продукта и последствиями, которые оно вызывает.
И если раньше меня печалило, что мне 14 лет, а я ни разу не пробовал колу, то потом я стал думать: «Ну и хорошо, что не попробовал. А то, кто знает, что было бы?». Я питаюсь, как и остальные члены моей семьи, просто исключив вредные для меня продукты.
Абсолютно все, что связано с применением физической силы, вызывает у меня проблемы. Например, мыть посуду я могу только в перчатках, но на деформированную кисть перчатка не налезает. Если делать это без перчаток, может быть какая-то нехорошая реакция на моющее средство, а кожа на руках будет сохнуть еще сильнее. Я не могу самостоятельно открыть бутылку воды, потому что из-за этого механического воздействия мои ладони травмируются. Я могу открыть не любую дверь. Если дверная ручка шарообразной формы, да ещё и с рельефом, травмы не избежать. Я не могу преодолевать большие расстояния пешком. Чем дольше я иду, тем сильнее это травмирует мои ноги. Даже небольшой выступ на асфальте может быть достаточным для появления раны на стопе. И так со многими действиями.
Но я не совсем беспомощный — с технологиями проблем нет. Я пишу, пользуюсь клавиатурой и мышкой. В работе мне приходится часто и быстро печатать большое количество текста, в том числе на телефоне. С этим я справляюсь.
Об отношении к болезни
Мое заболевание никогда не было для меня чем-то, что заставляло бы меня отчаиваться и падать духом. Скорее, я воспринимал его как само собой разумеющийся факт. Например, как тот, что солнце встает на востоке и заходит на западе.
Я осознаю свои ограничения. Тут вопрос не в том, осознаю ли я их, а в том, как я к ним отношусь. С детства в силу возраста и характера я переживал из-за этого не так сильно, как мог бы. А сейчас мне 26 лет, и я привык к этим ограничениям.
Многие мои сверстники водят автомобиль, а я нет. Когда мне было 18, меня это сильнее трогало, сейчас мне это глубоко индифферентно. Не вожу — и не вожу. Может, и к лучшему. Ни в кого не врежусь и не буду за это привлечен к ответственности. Хорошо, что не ем всякую химозу, зато у меня здоровый желудок. Какое-то такое отношение. С годами стало проще.
Понятно, что некоторые проявления болезни доставляют дискомфорт, но я отношусь к этому со смирением и осознанием того, что если что-то нельзя, то и ладно, живем дальше. С этим можно жить.
И главный вопрос «Как жить?». Отчаявшись? Не ставя перед собой никаких целей? Жить, как получится? Или, даже несмотря на заболевание, стараться чего-то достичь, кому-то помочь? Мне повезло, что сейчас не Средние века, и я вижу много возможностей для самореализации и социализации даже при наличии таких существенных отклонений, как у меня.
О друзьях, учебе и жизни в обществе
Никакого буллинга в мою сторону никогда не было, по крайней мере активного. Я учился в обычной средней общеобразовательной школе, потом поступил в университет. И везде у меня появлялись друзья — общения в моей жизни немало.
Я юрист, сейчас одновременно учусь в аспирантуре и прохожу стажировку в крупной компании. Все выходные и праздники заполняю дополнительной учебой.
Два моих главных хобби с детства — чтение книг и компьютерные игры.
Люблю читать психологов, один из моих любимых — Виктор Франкл. Его «Скажи жизни да» — это, можно сказать, моя настольная книга.
Из последних впечатливших меня книг, — «Игры, в которые играют люди» Эрика Берна. Однако чем старше я становлюсь, тем меньше художественной и больше профессиональной литературы я читаю.
Как я уже говорил, в обычной жизни я испытываю ограничения там, где у других людей их нет. Однако не могу сказать, что испытываю какую-то радость, когда у меня получается что-то, что получается у других. Обычно наоборот: я испытываю радость, когда я делаю что-то, что не могут другие. Вот тогда да! Во многом это касается профессиональных успехов.
О техническом прогрессе, который помогает людям-бабочкам
Кто знает: возможно, лет через 5-10 я и машину смогу водить. Например, еще 5 лет назад у меня не было никакой возможности поставить коронки на зубы. До 8 лет мои зубы в принципе лечить не хотели. «Мы не можем это лечить. Ваш рот плохо открывается. Мы боимся вас травмировать».
Был случай, когда в одной клинике врач выбежала из кабинета с криками: «Я не хочу в тюрьму!». Потому что мое заболевание не только про травмы кожи, но и про травмы слизистых оболочек.
Недавно фонд «Дети-бабочки» обучил врачей в клинике челюстно-лицевой хирургии имени Сеченова. И сейчас мне поставили на некоторые зубы коронки, появились технологии, которые это позволяют. Это та реальность, которая придает мне оптимизм. Технический прогресс на месте не стоит, и он работает на меня.
Однако я бы не согласился участвовать в клиническом исследовании нового препарата от своей болезни. Несмотря на все ограничения, которые у меня есть, я выбираю свою нынешнюю жизнь со всеми неудобствами, а не рискованные методы лечения.
Отдельно я хочу выразить благодарность фонду «Дети-бабочки». Он вносит существенный вклад в мою жизнь и в жизнь других людей с буллезным эпидермолизом — до него нам было намного сложнее.
Благодаря просветительской работе фонда, намного больше врачей знают, что есть такая болезнь. И значит, люди с таким диагнозом могут к ним прийти, быть понятыми и получить помощь.
Классификация буллёзного эпидермолиза
Буллёзный эпидермолиз делится на четыре основных типа, различающихся по уровню кожи, на котором образуются пузыри:
- простой буллезный эпидермолиз (ПБЭ) — в верхних слоях эпидермиса;
- пограничный буллезный эпидермолиз (ПоБЭ) — на уровне светлой пластинки (lamina lucida);
- дистрофический буллезный эпидермолиз (ДБЭ) — в верхней части сосочкового слоя дермы, ниже плотной пластинки (lamina densa);
- синдром Киндлера — разный уровень образования пузырей.
Для каждого типа характерна своя тяжесть проявлений и различные сочетания мутаций в разных генах. Типы мутаций определяют также характер наследования заболевания: аутосомно-доминантный или аутосомно-рецессивный. В настоящее время выявлено 18 генов, мутации в которых связаны с различными подтипами БЭ.
Клинические проявления БЭ
Буллёзный эпидермолиз проявляется уже во время родов: кожа ребёнка травмируется, когда он проходит через родовые пути. Обычно страдают нос, подбородок и пятки. В редких случаях болезнь дает о себе знать на 1-6 месяце жизни. С возрастом, в зависимости от типа БЭ, могут проявляться новые симптомы заболевания.
Основным клиническими проявлениями буллёзного эпидермолиза являются пузыри на коже, появляющиеся на местах трения, ушиба, давления, при повышении температуры тела, окружающей среды или спонтанно. Образование пузырей может возникать и на слизистых оболочках в любом органе. Чаще всего поражается слизистая оболочка полости рта, пищевода, кишечника, мочеполовой системы, слизистой глаз.
Простой буллёзный эпидермолиз
Современная классификация буллёзного эпидермолиза делит ПБЭ на 12 подтипов. Наиболее распространёнными подтипами ПБЭ являются: локализованный подтип (ранее тип Вебера-Коккейна), генерализованный подтип (ранее Доулинга-Меары или герпетиформный), генерализованный подтип другой (ранее Кёбнера), простой буллёзный эпидермолиз с пятнистой пигментацией.
Фенотип подтипов варьируется, пузыри могут появляться на кистях рук и стопах, а могут покрывать все тело. Обычно пузыри заживают без образования рубцов. В редких случаях присоединившаяся к множественным пузырям вторичная инфекция может привести к летальному исходу.
Наиболее распространённым подтипом простого БЭ является локализованный подтип. Обычно в семьях большое количество больных и заболевание встречается в нескольких поколениях. При этом подтипе пузыри локализуется на ладонях и подошвах. В раннем возрасте могут носить распространённый характер, но с возрастом проявления уменьшаются. Клинические проявления обостряются летом.
Самый тяжёлый вариант простого БЭ - генерализованный подтип Доулинга-Меары. Он характеризуется наличием пузырей или везикул, возникающих группами. Существует название «герпетиформный простой БЭ», поскольку некоторые повреждения похожи на те, что возникают при простом герпесе. Заболевание проявляется в момент рождения, степень тяжести у всех больных варьируется. При этом подтипе отмечается распространённый или сливной ладонно-подошвенный гиперкератоз, дистрофия ногтей, атрофическое рубцевание, милии, гипер- и гипопигментация и повреждение слизистых. Образование пузырей может принимать тяжёлую форму, иногда приводящую к смерти. Этот подтип может вызывать задержку роста и стеноз гортани.
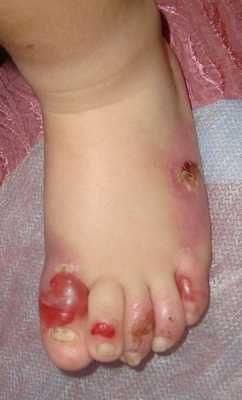
Пограничный буллёзный эпидермолиз
Пограничный БЭ также характеризуется хрупкостью кожи и слизистых оболочек, спонтанным появлением пузырей практически без травм. Одним из признаков является образование грануляционной ткани на определенных частях тела. Пузыри обычно заживают без существенных рубцов.
Пограничный тип БЭ делят на два основных подтипа, один из которых подразделяется ещё на шесть подтипов. Основные подтипы пограничного БЭ: подтип Херлитца (ранее летальный), подтип не-Херлитца (ранее генерализованный атрофический).
Подтип Херлитца наиболее тяжёлый генерализованный вариант пограничного БЭ. При нем высок риск преждевременной смерти. К типичным симптомам относятся образование множества пузырей, эрозий и атрофических рубцов кожи, ониходистрофия, приводящая к полной утрате ногтевых пластин и серьёзным рубцам ногтевых лож, милии, тяжёлое поражение мягких тканей в ротовой полости, гипоплазия эмали и тяжёлый кариес зубов. Патогномоничным симптомом является обильная грануляционная ткань, симметрично образующаяся вокруг рта, в средней части лица и вокруг носа, в верхней части спины, подмышечных впадинах и ногтевых валиках. Возможными системными осложнениями являются тяжёлая полиэтиологическая анемия, задержка роста, эрозии и стриктуры желудочно-кишечного тракта, поражение слизистых оболочек верхних дыхательных путей и мочеполового тракта, поражение почек, наружных оболочек глаза и в редких случаях поражение кистей рук. Смертность крайне высока, особенно в первые несколько лет жизни, в результате прекращения прибавки в весе, сепсиса, пневмонии или обструкции гортани и трахеи.
Подтип не-Херлитца проявляется образованием генерализованных пузырей, эрозий и корок на коже, атрофических рубцов, рубцовой алопеции («по мужскому типу»), дистрофией или потерей ногтей, гипоплазией эмали и кариесом.
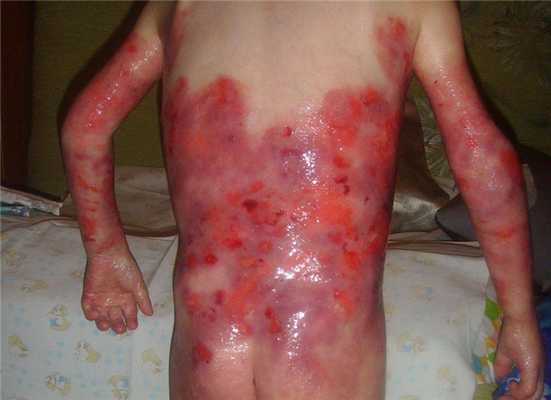
Дистрофичный буллезный эпидермолиз
Дистрофический БЭ делится на два главных подтипа в зависимости от наследования: доминантный дистрофический и рецессивный дистрофический (ДДБЭ и РДБЭ).
Доминантный дистрофический БЭ клинически характеризуется рецидивирующим образованием пузырей, милиями и атрофическим рубцеванием, особенно на конечностях, а также дистрофией и, в конечном итоге, утратой ногтей. У большинства пациентов поражение кожи является генерализованным. К внекожным проявлениям относятся осложнениями со стороны желудочно-кишечного тракта.
Рецессивный дистрофический БЭ делится на два подтипа: тяжёлый генерализованный подтип (ранее Аллопо-Сименса) и генерализованный другой подтип (ранее не-Аллопо-Сименса). Рецессивный дистрофический тяжёлый генерализованный подтип характеризуется генерализованным образованием пузырей, эрозиями, атрофическими рубцами, ониходистрофией и утратой ногтей, псевдосиндактилией пальцев рук и ног. Поражение кожи носит обширный и устойчивый к терапии характер. При рецессивном дистрофическом генерализованном другом подтипе характеризуется пузыри локализуются на руках, ногах, коленях и локтях, иногда на сгибах, на туловище.
При всех подтипах РДБЭ с возрастом развиваются контрактуры суставов локтей и коленей, кистей и стоп. Часто встречаются внекожные проявления: поражения желудочно-кишечного и мочеполового трактов, внешних оболочек глаза, хроническую анемию, остеопороз, задержку роста. У пациентов с РДБЭ высокий риск онкологических заболеваний, в частности образования агрессивных плоскоклеточных карцином.
Паллиативная медицинская помощь детям с буллезным эпидермолизом

Типы буллезного эпидермолиза, кожные и внекожные проявления заболевания, особенности ухода за новорожденным с БЭ и ведение таких детей мультидисциплинарной командой
В статье, вышедшей в третьем номере журнала Pallium за 2019 год, изложены современные представления о клинической картине и методах симптоматической терапии у детей с различными формами буллезного эпидермолиза. Особое внимание уделено ведению пациентов в периоде новорожденности. Представлены пилотные результаты добровольного анкетного опроса врачей по различным аспектам помощи детям с буллезным эпидермолизом. Благодарим редакцию Pallium за предоставленный материал.
Буллезный эпидермолиз (БЭ) - неизлечимое кожное заболевание, ограничивающее продолжительность жизни. Пациенты с БЭ отвечают всем критериям нуждаемости в паллиативной медицинской помощи, хотя не всегда и не всюду понимаются врачами как таковые. Тяжелый хронический характер их полиорганной патологии и отсутствие этиопатогенетического лечения
Читайте также: