Диагностика нейрофибромы по КТ, МРТ
Добавил пользователь Cypher Обновлено: 22.01.2026
Нейрофиброматоз имеет 2 типа. 1 тип - нейрофиброматоз реклингхаузена — наследственное заболевание, предрасполагающее к образованию опухолей. Частота: 1:2300 (самый распространенный нейрокожный синдром).
- Аутосомно-доминантный тип наследования
- Локус гена на хромосоме 17 (17ц 11)
- В 80% случаев причиной является новая мутация
- Форма нарушения вторичной нейруляции (дефект нейрональной пролиферации, дифференциации и гистогенеза) в течение 2-4-го месяцев беременности.
- Гистология: Нейрофибромы состоят из шванновских клеток, нейронов и коллагена в неорганизованном межклеточном матриксе
- В отличие от неврином (шванном) для них характерно отсутствие капсулы и более высокое содержание коллагена и эластина.
- Нейрофиброматоз также имеет экстрацеребралъные проявления:
- Плексиформные нейрофибромы головы и шеи
- Спинальные нейрофибромы.
Клинические проявления
- Пятна цвета «кофе с молоком»
- Кожные нейрофибромы
- Выпячивание глазного яблока, которое может быть обусловлено глиомой зрительного нерва
- Иногда пульсирующий экзофтальм
- Нейропсихические расстройства разной выраженности
- Иногда макроцефалия.
- По меньшей мере 5 пятен цвета «кофе с молоком» размером более 5 мм.
- Одна плексиформная нейрофиброма или две и более кожных/подкожных нейрофибромы.
- Пигментные пятна в подмышечной или паховой области.
- Дисплазия крыла клиновидной кости или длинных трубчатых костей.
- Односторонние или двусторонние глиомы зрительного нерва.
- Два или более узелка Лиша (гамартомы радужки).
- Отягощенный семейный анамнез.
- Нейрофиброматоз у детей может проявлятся нарушение слуха или снижением остроты зрения.
Какой метод диагностики нейрофироматоза I типа выбрать: МРТ, КТ, ангиографию
Метод выбора
Информативна ли МСКТ головы при болезни Реклингхаузена
- Дисплазия крыла клиновидной кости
- Дисплазия кости вдоль ламбдовидного шва
- Дефекты свода черепа.
Что покажут снимки МРТ головного мозга при нейрофироматозе I типа
- Глиомы переднего зрительного пути: двустороннее утолщение зрительного нерва (зрительного перекреста, зрительного пути) с минимальным контрастным усилением или без него
- Глиомы крыши среднего мозга: утолщение крыши среднего мозга и облитерация водопровода мозга с окклюзионной гидроцефалией
- Глиомы могут также встречаться в стволе головного мозга, мозжечке и полушариях большого мозга.
- Гиперинтенсивные очаги на Т2-ВИ в стволе мозга, внутренней капсуле и базальных ядрах, а также в валике мозолистого тела и белом веществе мозжечка
- Отсутствие контрастного усиления после введения КС
- Типично встречается в возрасте старше 3 лет
- Количество и размеры очагов увеличиваются до 10-12-летнего возраста
Вакуолизация миелина: почти никогда не встречается у больных в возрасте старше 20 лет
- Гиперинтенсивная зона на нативных Т1-ВИ (репаративные процессы)
- Нейрофибромы основания черепа и лица гиперинтенсивны по отношению к скелетной мышце на Т1- и Т2-ВИ
- Очаги могут накапливать КС
- При дисплазии крыла клиновидной кости височная доля пролабирует в глазницу.
Проводят ли ангиографию сосудов мозга при нейрофиброматозе
- Аневризмы головного мозга
- Другие сосудистые мальформации.,
Что хотел бы знать лечащий врач
- В зависимости от терапевтической значимости динамические исследования глиом или гидроцефалии
- Показать нейрофибромы.
Какие заболевания имеют симптомы, схожие с нейрофиброматозом I типа
Доброкачественный стеноз водопровода мозга:
- Расширение проксимального отдела водопро вода мозга
- Крыша среднего мозга истончена и смещена вверх.
Лечение
- Лечение гидроцефалии
- Удаление нейрофибром или глиом по показаниям (глиомы зрительного нерва удаляются только после подтверждения снижения остроты зрения или инфильтративного роста)
- Хирургическая пластика дефекта крыла клиновидной кости.
Врачи каких специальностей диагностируют и лечат нейрофиброматоз
- Невропатолог (определение неврологического дефицита)
- нейрохирург (при необходимости хирургическое вмешательство)
-врач-функциональной диагностики (дифференциальная диагностика с другими заболеваниями головного мозга)
Прогноз
Обычно благоприятный, перерождение в злокачественные опухоли бывает редко.
Возможные осложнения и последствия
Нейропсихические расстройства разной выраженности
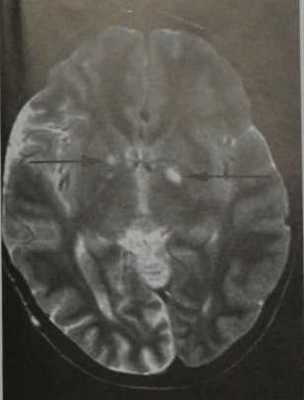
Нейрофиброматоз I типа. МРТ , Т2-Вив аксиальной плоскости. Множественные двусторонние гиперинтенсивные очаги во внутренней капсуле (вакуолизация миелина, стрелкой).
МРТ и КТ при нейрофиброматозе - что покажет

МРТ головного мозга, КТ головного мозга являются аппаратными методами диагностики нейрофиброматоза. Исходя из первичных симптомов, истории болезни, противопоказаний и диагностических целей, врач невролог назначает необходимые способы обследования. В большинстве случаев, МРТ головного мозга выбирается как первичный метод диагностики. Если результаты исследования носят тревожный или неясный характер, дополнительно назначается КТ головного мозга.
Что такое нейрофиброматоз
Нейрофиброматозы — это группа генетических расстройств, которые вызывают образование опухолей в нервной ткани. Эти опухоли могут развиваться в любой точке нервной системы, включая мозг, спинной мозг и нервы. Существует три типа нейрофиброматоза: нейрофиброматоз 1 типа, нейрофиброматоз 2 типа и шванноматоз. Тип 1 обычно диагностируется в детстве, в то время как тип 2 и шванноматоз обычно диагностируются в раннем взрослом возрасте. Опухоли при этих расстройствах обычно нераковые (доброкачественные), но в некоторых случаях могут стать злокачественными. Симптомы часто легкие. Однако осложнения нейрофиброматоза могут включать в себя потерю слуха, психоэмоциональные проблемы, проблемы с сердцем и кровеносными сосудами, потерю зрения и сильную боль.Лечение нейрофиброматоза направлено на стимуляцию здорового роста и развития детей, страдающих расстройством, и на раннее выявление осложнений. В том случае, когда нейрофиброматоз вызывает крупные опухоли или опухоли, которые давят на нерв, операция может уменьшить симптомы. Для некоторых пациентов назначаются специализированные методы лечения, такие как стереотаксическая радиохирургия или прием медикаментозных препаратов для борьбы с болью.
Симптомы нейрофиброматоза
Существует три типа нейрофиброматоза, каждый из которых имеет разные признаки и симптомы.
Симптомы нейрофиброматоза 1 типа
Нейрофиброматоз 1 типа обычно диагностируется в детстве. Признаки часто заметны при рождении или вскоре после него, и почти всегда к 10 годам. Симптомы варьируются по степени тяжести и включают в себя:
плоские светло-коричневые пятна на коже. Эти безобидные пятна распространены у многих пациентов. Наличие более шести пораженных областей предполагает заболеваний. Обычно они присутствуют при рождении или появляются в первые годы жизни. Во взрослом возрасте новые пятна перестают появляться
- веснушки в подмышках или в области паха. Веснушки обычно появляется в возрасте от 3 до 5 лет
- крошечные узелки на радужной оболочке глаза (узелки Лиша). Образование узелков не влияет на зрение
- мягкие шишки размером с горох на коже. Эти доброкачественные опухоли обычно развиваются в коже или под кожей, но также могут расти внутри тела. Новообразование включает в себя много нервов (оргсиформная нейрофиброма). Плексиформные нейрофибромы, расположенные на лице, могут привести к значительному ухудшению внешности и серьезному поражению кожи лица. С возрастом состояние может ухудшаться
- деформации костей. Аномальное развитие и дефицит минеральной плотности костей могут вызвать деформации, такие как изогнутый позвоночник (сколиоз) или изогнутая голень
- опухоль на зрительном нерве (глиома зрительного нерва). Эти опухоли обычно появляются в возрасте 3 лет, редко в позднем детстве и подростковом возрасте и крайне редко у взрослых пациентов
- нарушения в обучении. Нарушение навыков мышления распространено у детей. Часто развивается специфическая неспособность к обучению. Также распространены синдром дефицита внимания/гиперактивности (СДВГ) и задержка речи
- увеличенный размер головы. Дети с нейрофиброматозом 1, как правило, имеют увеличенный размер головы из-за увеличения объема мозга
- замедленный рост и развитие.
Симптомы нейрофиброматоза 2 типа
Нейрофиброматоз 2 типа встречается гораздо реже. Симптомы обычно являются результатом развития доброкачественных, медленно растущих опухолей в обоих ушах (акустические неврозы), которые могут привести к потере слуха. Также известные как вестибулярные шванномы, эти опухоли поражают нерв, который несет звуковую и балансовую информацию от внутреннего уха до мозга. Симптомы обычно появляются в позднем подростковом и раннем взрослом возрасте и могут варьироваться по степени тяжести:
- постепенная потеря слуха
- звон в ушах
- плохой баланс
- головные боли.
Иногда нейрофиброматоз 2 может привести к росту шванном в других нервах, включая черепные, спинальные, зрительные (оптические) и периферические нервы. У пациентов также могут развиться другие доброкачественные опухоли. Симптомы при появлении дополнительных опухолей:
- онемение и слабость в руках или ногах
- трудности баланса
- отек лица
- проблемы со зрением или катаракта
- судороги
- головная боль.
Симптомы шванноматоза
Этот редкий тип нейрофиброматоза обычно поражает пациентов после 20 лет. Симптомы обычно появляются в возрасте от 25 до 30 лет. Шванноматоз вызывает развитие опухолей на черепных, спинномозговых и периферических нервах, но редко на нерве, который передает звуковую и балансовую информацию от внутреннего уха к головному мозга. Опухоли обычно не растут на обоих слуховых нервах, поэтому пациенты с шванноматозом не испытывают такой же потери слуха, как при нейрофиброматозе 2 типа. Симптомы шванноматоза:
- хроническая боль, которая может возникнуть в любом месте тела
- онемение или слабость в различных частях тела
- мышечная атрофия.
Причины нейрофиброматоза
Нейрофиброматоз вызван генетическими дефектами (мутациями), которые либо наследуются, либо происходят спонтанно при зачатии. Конкретные типы нейрофиброматоза зависят от ряда генов:
- нейрофиброматоз 1 типа. Аномальный ген располагается расположен на 17-й хромосоме. Этот ген производит белок под названием нейрофибромин, который помогает регулировать рост клеток. Мутировавший ген вызывает потерю нейрофибромина, что позволяет клеткам расти бесконтрольно
- нейрофиброматоз 2 типа. Пораженный ген расположен на хромосоме 22 и производит белок под названием мерлин (также называемый шванномин), который подавляет образование и рост опухолей. Мутировавший ген вызывает потерю мерлина, что приводит к неконтролируемому росту клеток
- шванноматоз. Мутации генов SMARCB1 и LZTR1, которые подавляют опухоли, связаны с этим типом нейрофиброматоза.
Факторы риска
Самым большим фактором риска нейрофиброматоза является соответствующая наследственность. Около половины пациентов, у которых есть заболевание 1 и 2 типа, унаследовали болезнь от пострадавшего родителя.
Нейрофиброматоз 1 и 2 являются аутосомно-доминантными расстройствами, что означает 50-процентную вероятность наследования аномальных генов.
Модель наследования шванноматоза менее ясна. По оценкам исследователей, риск наследования шванноматоза от пострадавшего родителя составляет около 15%.
Осложнения
Осложнения нейрофиброматоза варьируются даже в одном и том же семействе. Как правило, осложнения возникают из-за опухолей, которые влияют на нервную ткань или оказывают давление на внутренние органы.
Осложнения нейрофиброматоза 1 типа
Основные осложнения включают в себя:
- неврологические проблемы. Трудности в обучении и мышлении являются наиболее распространенными неврологическими проблемами, связанными с нейрофиброматозом. Необычные осложнения включают в себя эпилепсию и накопление избыточной жидкости в мозге
- проблемы с внешностью. Видимые признаки нейрофиброматоза, такие как обширные темные пятна, многие нейрофибромы в области лица, могут вызвать беспокойство и эмоциональный стресс, даже если они не являются серьезными с медицинской точки зрения
- проблемы со скелетом. У некоторых детей аномально сформированные кости, что может привести к переломам, которые иногда не заживают. Нейрофиброматоз 1 может вызвать кривизну позвоночника (сколиоз), требующую хирургического вмешательства. Заболевание также связано со снижением минеральной плотности костной ткани, что увеличивает риск остеопороза
- проблемы со зрением. У пациентов может развиться глиома зрительного нерва
- гормональные сбои. Гормональные изменения, связанные с половым созреванием или беременностью, могут привести к увеличению нейрофибром. Большинство женщин, страдающих нейрофиброматозом, имеют здоровую беременность, но, вероятно, нуждаются в мониторинге со стороны акушера
- сердечно-сосудистые проблемы. Пациенты имеют повышенный риск высокого артериального давления, кроме того могут развиться аномалии кровеносных сосудов
- проблемы с дыханием. Редко оргсиформные нейрофибромы могут оказывать давление на дыхательные пути
- рак. По оценкам, у 3%-5% пациентов с нейрофиброматозом развиваются раковые опухоли. Обычно они возникают из-за нейрофибром под кожей или из плексиформных нейрофибром. Пациенты с нейрофиброматозом 1 также имеют более высокий риск других форм рака, таких как рак молочной железы, лейкемия, колоректальный рак, опухоли головного мозга и некоторые виды рака мягких тканей. Женщинам рекомендуется ранний скрининг на рак молочной железы
- опухоль надпочечников (феохромоцитома). Эта доброкачественная опухоль выделяет гормоны, которые повышают артериальное давление. Для удаления фиохромоцитомы назначается специальная операция.
Осложнения нейрофиброматоза 2 типа
- частичная или полная глухота
- повреждение лицевого нерва
- проблемы со зрением
- небольшие доброкачественные опухоли кожи (шванномы)
- слабость или онемение в конечностях
- множественные доброкачественные опухоли головного мозга или опухоли позвоночника (менингиомы), требующие частых операций.
Осложнения шванноматоза
Боль, вызванная шванноматозом, может быть изнурительной. Зачастую пациентам требуется хирургическое вмешательство.
МРТ и КТ в диагностике нейрофиброматоза
Обследование начинается с обзора личной и семейной истории болезни и физического осмотра пациента. Также врач обследует кожу на наличие пятен и иных поражений. Дополнительно для диагностики нейрофиброматоза обоих типов или шванноматоза назначаются следующие процедуры:
- осмотр глаз. Офтальмолог может обнаружить узелки Лиша, катаракту и потерю зрения
- исследование слуха и равновесия. Пациенту назначается аудиометрия и электронистагмография - обследование, при котором используются специальные электроды для записи движений глаз. Кроме того, измеряются электрические сигналы, которые передают звук от внутреннего уха к мозгу (звуковой сигнал ствола мозга)
- Компьютерная томография или МРТ позволяют выявление аномалии костей, опухоли головного или спинного мозга и другие очень мелкие опухоли. МРТ головного мозга может быть использована для диагностики глиомы зрительного нерва. Данные процедуры также часто используются для мониторинга нейрофиброматоза и шванноматоза
- генетические исследования. Данные исследования могут быть выполнены во время беременности до рождения ребенка. Генетические тесты не всегда идентифицируют шванноматоз, потому что другие неизвестные гены могут быть связаны с расстройством. Тем не менее, некоторые женщины выбирают генетическое тестирование SMARCB1 и LZTR1 перед рождением детей.
Для диагностики у пациента должно быть по крайней мере два признака состояния. Если в наличии только один признак и нет семейного анамнеза нейрофиброматоза, то пациенту назначается регулярное наблюдение для выявления каких-либо дополнительных признаков. Диагноз нейрофиброматоза 1 обычно ставится к 4 годам.
Разница между МРТ и КТ
Компьютерная томография и магнитно-резонансная томография используются для получения изображений органов и тканей в трехмерной проекции. Разница в томографии заключается в том, что при МРТ используются радиоволны, а при КТ - рентгеновские лучи для получения изображения. Хотя оба томографических метода имеют высокую информативность, есть диагностические различия, которые могут сделать каждый из них более подходящим вариантом в зависимости от обстоятельств.
КТ-сканирование более быстрая форма диагностики, чем магнитно-резонансная томография, и обычно используется для экстренного обследования. При использовании как КТ, так и МРТ существуют определенные риски. Они зависят от типа визуализации, а также от способа ее проведения. К рискам КТ относятся: вред для внутриутробного ребенка при беременности и доза радиации. Риски МРТ включают реакции на металлические импланты в теле из-за сильного магнитного поля, громкий шум от аппарата, повышенная, клаустрофобические риски.
Если врачу нужны более детальные изображения мягких тканей, связок или органов высоким состоянием воды, обычно назначается магнитно-резонансная томография. Если необходимо получить хорошие изображение костных структур и полых органов, обычно рекомендуется компьютерная томография.
Нейрофиброматозы
Нейрофиброматозы - наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое - проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10

Общие сведения
Нейрофибромы - доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз - болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом - 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.
Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых - нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) - супрессора опухолевого роста. Тип наследования - аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора - частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи - пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз - факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом - гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента - гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет - 41%, с 10 до 19 лет - 85%, после 20 лет - 95%.
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко - деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы - подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак - появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
- МРТ, КТ. Визуализационные методы исследований позволяют определить наличие нейрофибром в головном мозге, позвоночнике, внутренних органах. Двусторонние невриномы 7 пары черепных нервов являются диагностическим критерием нейрофиброматоза II типа. Часто выявляются глиомы, шванномы, менингиомы. Для I типа свойственно развитие плексиформных и обычных новообразований, глиом.
- Рентгенография костей скелета. Диагностическая процедура выполняется с целью подтверждения и оценки тяжести сколиоза, костных атрофий и гипертрофий, локальных утолщений и эрозийных поражений костных структур. При большинстве типов болезни наблюдается истончение кортикального слоя, ложные суставы, дисплазии крыльев клиновидной кости, дугообразное искривление большеберцовой и малоберцовой костей, кисты длинных костей.
- Офтальмологическое исследование. Нейрофиброматоз типа 1 сопровождается плексиформной нейрофибромой век, меланоцитарными гамартомами радужки, глиомой оптических нервных волокон, астроцитарной гамартомой сетчатки, утолщением роговичных нервов, конъюнктивальной нейрофибромой, ишемическими поражениями венул сетчатки. Патогномоничный признак - пятна светлого оттенка на глазном дне и радужке (гамартомы). При 2 типе диагностируется задняя субкапсулярная катаракта, помутнение хрусталика.
- Аудиологическое исследование. При опухолевом поражении слуховых нервов и жалобах на нарастающую глухоту (тугоухость) выполняется электрокохлеография и импедансометрия. Результаты указывают на снижение остроты слуха, наличие слуховой нейропатии, определяют причину и локализацию нарушения.
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями - малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
1. Нейрофиброматоз: этиология, патогенез, лечение/ Скаварская Е.А.// Международный журнал педиатрии, акушерства и гинекологии - 2014 - Т.5, №2.
2. Клинико-диагностические аспекты нейрофиброматоза/ Попова А.А.// Университетская медицина Урала - 2016 - №2.
3. Нейрофиброматоз первого типа (болезнь Реклингхаузена)/ Н.А. Шнайдер, А.И. Горелов// Сибирское медицинское обозрение - 2007.
Факоматозы и их диагностика
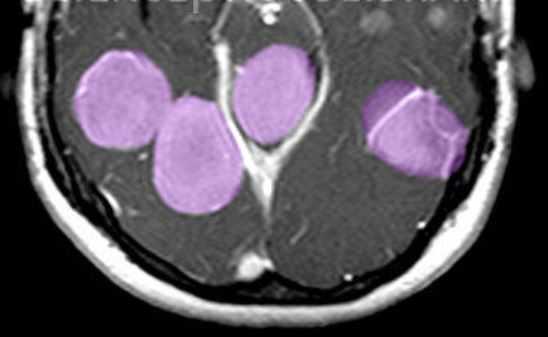
МРТ головного мозга. Т1-взвешенная МРТ с контрастированием. Множественные менингиомы при нейрофиброматозе 2 типа. Цветовая обработка изображения.
Факоматозы (нейрокожные нарушения) - это группа врожденных дисплазий эмбриональной эктодермы (кожи, центральной и переферической нервной систем, глаза) и эндодермы (эпителиальной выстилки желудочно-кишечного тракта).
К факоматозам относят:
- Нейрофиброматоз тип I (болезнь Реклингхаузена)
- Нейрофиброматоз тип II
- Туберозный склероз (синдром Бурневиля)
- Болезнь Гиппель-Линдау
- Синдром Стурге-Вебера
- Редкие заболевания - синдром Клиппеля-Треноне-Вебера, синдром Протея, синдром Ослера-Рандю-Вебера, синдром Вайбурна-Масона, болезнь Фабри, синдром кожной гемангиомы, атаксия-телеангиэктазия (синдром Луи-Бара), менингоангиоматоз (синдром Ульмана), нейрокожный меланоз, гипомеланоз Ито, синдром эпидермального невуса, базальноклеточный невус.
Все перечисленные заболевания выявляются при МРТ головного мозга.
Нейрофиброматоз I типа (болезнь Реклингхаузена) имеет аутосомно - доминантный тип наследования (50%), сцепленный с 17 парой хромосом, или с её спонтанными мутациями. Частота заболевания составляет один на 4000 населения. Диагностическими критериями служат (National Institutes of Health, 1988) не менее 6 кожных пятен (макулы цвета “кофе с молоком” размером не менее 5 мм в пре- и 15 мм в послепубертатном периоде), и не менее 2 любых нейрофибром, либо одна плексиформная (подкожная) нейрофиброма, множественные веснушки в подмышечных и паховых областях, костные дисплазии (истончение кортикального вещества длинных трубчатых костей с коксартрозом или без него, дисплазия клиновидной кости ), двухсторонние глиомы зрительных нервов, 2 или более пигментированные гамартомы радужной оболочки глаза (узлы Лиша) и наличие ближайшего родственника с этим заболеванием. Для постановки диагноза достаточно наличия 2 из перечисленных критериев. Кроме того, характерны следующие сопутствующие патологии: невриномы, кожные нейрофибромы, макроцефалия, астроцитомы, множественные менингиомы, кифосколиоз, саркома Юинга, сирингомиелия. Из опухолей головного мозга наиболее часто (в 5-15% случаев НФ I) встречается пилоцитарная астроцитома зрительного нерва и тракта . На МРТ астроцитома видна в виде утолщения зрительного нерва. Образование изоинтенсивно или немного гипоинтенсивно на Т1-взвешенных МРТ и гиперинтенсивно на Т2-взвешенных МРТ. Среди других локализаций астроцитома может быть в стволе, гипоталамусе, III желудочке. Редко обнаруживаются анапластические астроцитомы полушарий и мозжечка. У детей при НФ I встречаются очаги , напоминающие гамартомы. Они выявляются в ножках мозга, мосте, бледном шаре, среднем мозге, зрительном бугре, продолговатом мозге, реже белом веществе полушарий и мозжечка. На Т2-взвешенных МРТ очаги слегка гиперинтенсивны, на Т1-взвешенных МРТ изоинтенсивны белому веществу, за исключением бледного шара, где они тоже чуть гиперинтенсивны. Они не превышают 15 мм в размерах, не вызывают масс-эффекта и не контрастируются. Предположительно, очаги представляют собой участки изменённого миелина.
Третьим образованием диагностируемым при нейрофиброматозе I типа является нейрофиброма, обычно расподоженная в орбите и распространяющаяся в полость черепа и на кавернозный синус. Нейрофиброма слегка гиперинтенсивнее мышцы на Т1-взвешенных МРТ и выраженно гиперинтенсивнее на Т2-взвешенных МРТ. Кистозный компонент опухоли может давать сниженный сигнал в ее центре на Т1-взвешенных томограммах. При нейрофиброматозе I типа встречается также эктазия твердой мозговой оболочки в расширенном слуховом канале и стеноз водопровода. Эктазию важно не путать с невриномой. Стеноз водопровода не связан с опухолевой компрессией, но приводит к гидроцефалии.
Нейрофиброматоз II типа также имеет аутосомно-доминантный тип наследования, но сцепленный с 22 парой хромосом. Его частота составляет один на 100 тыс. населения. Диагностическими критериями служат (National Institutes of Health, 1988) двухсторонние невриномы слуховых нервов, выявляемые на КТ или МРТ, либо сочетание наследственной предрасположенности (наличие двухсторонних неврином у ближайшего родственника) с односторонней невриномой или двумя другими типичными опухолями (плексиформная нейрофиброма, менингиома, глиома, невринома любой локализации) плюс кожные пятна. В отличие от НФ I кожные пятна единичные и не служат главным критерием, а опухолевое поражение ассоциируется не с астроцитомами, а с невриномами и менингиомама. Сопутствующими патологиями являются менингоангиоматоз, глиальные узлы, эпендимальные эктопии, гипертрофический глиоз зрительного нерва, сирингомиелия, комплекс Арнольда-Киари. Типичная невринома развивается из шванновской оболочки слуховых нервов (VIII пара), обычно с обеих сторон , реже тройничного нерва или других. При МРТ невриномы гипо- или изоинтенсивны белому веществу на Т1-взвешенных МРТ и изо- или гиперинтенсивны на Т2-взвешенных МРТ. Хорошо усиливаются гадолинием. Менингиомы, как правило, сопутствуют невриномам. Локализация не отличается от случаев не связанных с нейрофиброматозом, но встречается также нетипичное поражение сосудистого сплетения. Картина менингиом при нейрофиброматозе II типа имеет все типичные признаки.
Туберозный склероз (синдром Бурневиля) встречается реже нейрофиброматоза. Его частота по данным литературы составляет около одного на 180 тыс. населения. От 20 до 40% случаев туберозного склероза унаследованы по аутосомно- доминантному типу, остальные возникли вследствии мутаций предположительно 9 и 11 пар хромосом (тип 1), либо 19 пары (тип 2). Поражение может затрагивать практически любые органы. Патогномоничными поражениями ЦНС являются корковые узлы в головном мозге и множественные субэпендимальные глиальные узлы , а также внутрижелудочковая гигантоклеточная астроцитома, встречаются сопутствующие аномалии - агенезия мозолистого тела, пахигирия, аневризмы. Характерны дерматологические проявления в виде множественных ангиофибром лица в форме «бабочки», бледные пятна на лице и груди, фибромы кожи, под ногтями и сетчатке глаза. Из других проявлений встречаются множественные ангиолипомы почек и печени, рабдомиомы сердца, лимфангиоматоз лёгких, костные склеротические и кистозные изменения. Диагноз туберозного склероза ставится при наличии у пациента 2 из перечисленных характерных признаков.
Корковые узлы - самое частое проявление туберозного склероза. Они расположены в коре головного мозга, деформируют её, захватывают прилегающее белое вещество и подвергаются кальцификации . При МРТ узлы изоинтенсивны серому веществу на Т1-взвешенных МРТ и чуть гиперинтенсивнее его на Т2-взвешенных. Контрастирование наблюдается в 5% случаев. В белом веществе обнаруживаются тяжи , отходящие радиально от желудочков. Корковые узлы и тяжи нередко называют «гамартомами», хотя они представляют собой скорее демиелинизацию и кальцификацию, чем истинную гетеротопию.
Туберозный склероз. Гамартомы. КТ, Т2-зависимая МРТ и FLAIR
Субэпендимальные, то есть проецирующиеся в желудочек, но растущие со стороны паренхимы мозга, узлы чаще расположены рядом с хвостатым ядром или гипоталамической бороздой сразу за отверстием Монро, реже в области III, IV желудочков и Сильвиева водопровода. На Т2-взвешенных томограммах субэпендимальные узлы умеренно гиперинтенсивны и часто содержат кальцинаты . От астроцитом их отличает не столь яркий сигнал и меньшие размеры. Контрастирование при введении препаратов гадолиния иногда наблюдается и в субэпендимальных узлах , и всегда в астроцитомах.
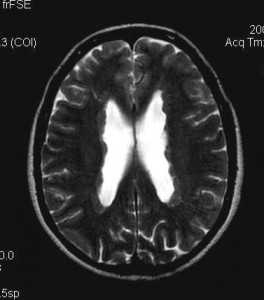
Туберозный склероз. Субэпендимальные узлы. Аксиальная Т-зависимая МРТ.
Болезнь Гиппеля - Линдау представляет собой симптомокомплекс, состоящий из гемангиобластом ЦНС (40% случаев) и сетчатки глаза (45%), и висцеральных проявлений в виде кист почек (75%), печени и поджелудочной железы. Диагноз ставится при наличии двух и более гемангиобластом или одной гемангиобластомы и висцеральных изменений, либо только висцеральных проявлениий при наличии семейной наследственности. Болезнь Гиппеля-Линдау в 20% случаев имеет врожденное семейное происхождение с аутосомно-доминантным типом наследования, в остальных связана с мутацией 3 пары хромосом. Частота примерно 1 случай на 36 тыс. населения. Кроме гемангиобластом и характерных висцеральных изменений при болезни Гиппель - Линдау встречается сопутствующие патологии: карцинома почки (более четверти наблюдений) и поджелудочной железы, феохромоцитома (около 10%, часто двухсторонняя), рабдомиома сердца, кисты лёгких, эпидидимит.
Гемангиобластомы при болезни Гиппеля - Линдау всегда множественные и примерно в половине случаев локализуются в мозжечке, реже стволе, спинном мозге и полушариях. При мозжечковой локализации опухоль чаще расположена поверхностно. При МРТ определяется неоднородный узел, гиперинтенсивный на Т2-взвешенных и изо- или гипоинтенсивный на Т1-взвешенных МРТ. В ряде случаев визуализируются патологически расширенные сосуды, имеющие характерное отсутствие сигнала. Кальцификации узлов не наблюдается. Гемангиобластомы хорошо контрастируются гадолинием. Чисто солидные гемангиобластомы наблюдаются только в 10% случаев. Окружающая узел киста гиперинтенсивна на томограммах обоих типов взвешенности, так как содержит примесь белка.
Синдром Стурге - Вебера ненаследуемое заболевание, ее морфологическим субстратом является ангиоматоз, связанный с тем, что сохраняются синусоидальные эмбриональные сосуды. Таким образом, синдром Стурге - Вебера представляет собой аномалию развития в “чистом” виде. Артериальный и венозный ангиоматоз приводят к избыточной васкуляризации оболочек мозга, кальцификации оболочечных артерий. Поражаются мягкие мозговые оболочки, обычно затылочной доли, причем с одной стороны (75% случаев). Кора мозга над ангиомой атрофируется и кальцифицируется . Нередко выявляется патологически расширенная кортикальная вена. Описано также увеличение сосудистого сплетения, гемиатрофия мозга на стороне поражения, ускорение и нарушение миелинизации, мегалэнцефалия и гидроцефалия. В постановке диагноза помогает наличие невуса кожи лба, который расположен по ходу первой ветви тройничного нерва, с той же стороны, что и очаг в мозге. Из других проявлений заболевания встречаются костные - ипсилатеральная гипертрофия черепа и синусов, глазные - ипсилатеральный экзофтальм, глаукома (30%), колобома радужной оболочки, гемангиома сосудистого сплетения глаза, висцеральные - ангиоматоз щитовидной железы, лёгких, поджелудочной железы, печени, почек, кишечника. Клинически синдром проявляется контрлатеральным гемипарезом, гомонимной гемианопией, судорогами (80% случаев) и умственной отсталостью (60%). При МРТ выявляется хорошо контрастирующийся ангиоматозный клубок сосудов, утолщенная оболочка, расширенная кортикальная вена и, иногда, расширенное сосудистое сплетение.
МРТ СПб дает место выбора выполнения МРТ головного мозга. При МРТ в СПб мы выступаем за комплексный подход к диагностике факоматозов с исследованием всех их проявлений. Обычно факоматозы лучше видны в высоких полях, но многие, особенно, опухоли видны и в низкопольных открытых МРТ.
Нейрофиброматоз II типа, определение 18 мутаций в гене NF2 в Барнауле
Исследование позволяет выявить 18 мутаций в гене NF2, которые приводят к развитию нейрофиброматоза II типа. У пациентов с таким заболеванием нарушены функции белка нейрофибромина-2, который подавляет рост опухолей. Анализ рекомендуется при подозрении на нейрофиброматоз II типа для подтверждения диагноза.
Приём и исследование биоматериала
Когда нужно сдавать анализ Нейрофиброматоз II типа, определение 18 мутаций в гене NF2?
Основные показания к проведению исследования:
- подозрение на нейрофиброматоз II типа;
- диагностика причин нарушения слуха;
- поиск причин развития множественных кожных опухолей, а также новообразований в нервной системе, головном и спинном мозге.
Подробное описание исследования
Нейрофиброматоз II типа
Нейрофиброматоз II типа — редкое генетическое заболевание, при котором в мягких тканях, внутренних органах, нервной системе и на коже образуются множественные доброкачественные опухоли.
Нейрофиброматоз возникает из-за мутации в гене NF2. Этот ген кодирует белок-супрессор нейрофибромин-2, подавляющий рост опухолей.
Заболевание встречается приблизительно у 1 из 50 000 человек.
Тяжесть заболевания зависит от вида мутации и её влияния на функции белка-супрессора.
НФ2 наследуется аутосомно-доминантно. Это значит, что, если один из родителей болен нейрофиброматозом II типа, вероятность рождения ребёнка с этой патологией составит 50%.
Признаки и симптомы
Признаки нейрофиброматоза II типа могут проявиться как с рождения, так и в подростковом возрасте. Как правило, дебют болезни происходит до 30 лет.
Основные признаки нейрофиброматоза II типа:
- нарушения слуха и зрения;
- коричневые пятна и многочисленные новообразования (нейрофибромы) на коже;
- опухоли слухового нерва (вестибулярные шванномы) в возрасте до 30 лет;
- многочисленные опухоли нервной системы, головного и спинного мозга;
- в некоторых случаях — костные деформации.
Пациенты также могут жаловаться на головную боль и эпилептические приступы.
В 10% случаев доброкачественные опухоли могут стать злокачественными.
Диагностика и лечение нейрофиброматоза II типа
При подозрении на нейрофиброматоз пациенту могут назначить КТ и МРТ головного мозга, обследование у офтальмолога. Для подтверждения диагноза проводят анализ гена NF2 на наличие мутаций.
Основной метод лечения нейрофиброматоза II типа — хирургическое удаление опухолей, радио- и химиотерапия, а также различные сочетания этих методов. При ранней диагностике и своевременном лечении прогноз для пациента благоприятный.
Читайте также:
- Витаминные сборы от гипертонии. Шиповник, боярышник и калина
- Схема пути MAPK в реакции эпителия кишечника на бактерии
- Асцит брюшной полости при онкологии: симптомы, жидкость в животе, водянка при раке
- Поля зрения, световосприятие при ртутной интоксикации
- Хирургическая обработка ран роговицы и склеры с выпадением радужки. Методика