Дифференциальный диагноз ахондроплазии у плода. Синдром акрофациального дизостоза - синдром Нагера
Добавил пользователь Alex Обновлено: 28.01.2026
Синдром Тричера Коллинза - это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
МКБ-10
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
Причины
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная - в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности - дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича - 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири - 2007 - №3.
Хондродисплазия
Хондродисплазии — это общее название для группы наследственных заболеваний, вызванных нарушением образования или окостенения хрящевой ткани. Они возникают в результате генных мутаций, наследуются как по аутосомно-доминантному, так и по аутосомно-рецессивному типу. Патологии проявляются низкорослостью, разнообразными укорочениями и деформациями конечностей, поражением суставов. Для диагностики необходимы данные рентгенографии или компьютерной томографии, результаты генетического исследования. Лечение направлено на устранение или коррекцию существующих нарушений, для чего применяются ортопедические операции, ортезы, методы ЛФК и физиотерапии.
На сегодня известно более 100 вариантов хондродисплазии, многие из которых схожи между собой по клинической картине, поэтому постановка точного диагноза, как правило, затруднена. Заболевания не теряют своей актуальности, поскольку сопровождаются грубыми деформациями опорно-двигательного аппарата, что чревато инвалидностью, резким ухудшением качества жизни, некоторые имеют высокий показатель летальности в раннем возрасте. Достоверные статистические данные распространенности отсутствуют.
Причины хондродисплазии
Патология связана с точечными мутациями генов (COL2A1, SEDL, COMP и т.д.), отвечающих за образование белков хрящевого матрикса или некоторых регуляторных пептидов. Большинство типов хондродисплазии имеют аутосомно-доминантный тип наследования — при наличии генного дефекта у одного из родителей риск рождения больного ребенка равен 50%. Некоторые формы отличаются аутосомно-рецессивным механизмом передачи, для которого оба родителя должны быть носителями мутантного гена.
При большинстве типов хондродисплазий выявляется первичный дефект белков: разных типов коллагена, матрилина, олигомерного протеина. Все они в норме составляют хрящевую ткань, поэтому при их патологическом строении наблюдаются дефекты структуры, избыточное или недостаточное развитие хрящей. Нарушения также происходят на этапе образования регуляторных белков, к которым относят тирозинкиназный рецептор, факторы транскрипции, ферменты процессинга РНК.
Типичные деформации и укорочение конечностей часто сочетаются с расстройствами энхондрального окостенения, при котором определяется неструктурное беспорядочное расположение хондроцитов, вызванное дефектами окислительного фосфорилирования. Ряд форм болезни проявляются образованием костных выступов (экзостозов). Патогенетические механизмы многих типов хондродисплазий до сих пор точно не изучены.
Классификация
В современной генетике принят комплексный подход к систематизации хондродисплазий, учитывающий этиопатогенетические, клинико-рентгенологические особенности. Международная рабочая группа по хондродисплазиям представила вариант классификации из 17 разделов согласно типу генной мутации. В то же время в практической медицине актуально деление болезни с учетом характера поражения хрящевой ткани, которое включает 3 категории:
- Эпифизарные дисплазии. Сюда относят поражения суставного хряща, дисплазии хрящевой ткани собственно эпифиза. Основные представители группы: болезнь Волкова, спондилоэпифизарная дисплазия, псевдоахондроплазия, болезнь Фэрбанка.
- Физарные дисплазии. Характеризуются нарушениями формирования ростковой эпифизарной пластинки. Эта категория включает ахондроплазию (хондродистрофию), гипохондроплазию, экзостозную хондродисплазию.
- Метафизарные дисплазии. Эта группа патологий возникает вследствие задержки энхондрального роста на фоне неправильного окостенения хрящей метафизов. Они наименее изучены, описано лишь несколько вариантов: дисхондроплазия (болезнь Олье), хондродисплазии типа Янсена, Шмида, Мак-Кьюзика.
Симптомы хондродисплазии
Типичным признаком любой формы болезни является низкий рост (карликовость), связанный с нарушениями удлинения трубчатых скелетных костей. Рост взрослых пациентов составляет около 1,2-1,3 м. Карликовость сопровождается диспропорциями тела: при относительно нормальных размерах туловища у таких больных короткие конечности, крупный череп. При раннем закрытии черепных швов происходит деформация головы.
Хондродисплазии имеют множество симптомов, зависящих от конкретного варианта патологии. У страдающих ахондроплазией большой лоб, седловидный нос, выраженный прогиб в пояснице (лордоз), ноги приобретают саблевидную форму. Для дистрофической дисплазии характерно удлинение больших пальцев кистей рук, эквиноварусная косолапость. При эпифизарных дисплазиях отмечаются укорочение дистальных фаланг пальцев, вывихи тазобедренных суставов.
Помимо симптомов со стороны опорно-двигательной системы диагностируются множественные соматические признаки хондродисплазий. Изменения кожи и подкожной клетчатки проявляются пигментными пятнами, крупными липомами, гемангиомами, на ладонях и стопах зачастую формируются большие очаги гиперкератоза. У пациентов могут возникать гепатоспленомегалия, разнообразные поражения глаз: дистрофия хрусталика, астигматизм, миопия.
Часть хондродисплазий относят к летальным мутациям, при их наличии лишь немногие пациенты доживают до взрослого возраста. В эту категорию выделяют некоторые типы ахондрогенеза, танатофорную дисплазию, капомелическую дисплазию, ризомелическую форму точечной хондродисплазии. К нелетальным последствиям заболевания относят контрактуры суставов, деформации конечностей, грубые искривления позвоночника по типу кифосколиозов или лордозов.
Поскольку поражение костно-хрящевых структур обычно затрагивает позвоночник, у больных есть риск неврологических нарушений. Они обусловлены сдавлением спинного мозга при сужении позвоночного канала либо при стенозе большого затылочного отверстия. Осложнения включают нарушения чувствительности, снижение тонуса скелетной мускулатуры, тазовые расстройства. Вследствие деформации грудной клетки нередко нарастает дыхательная недостаточность.
Генные мутации могут затрагивать не только белки хрящевой ткани, но и компоненты соединительнотканных волокон другой локализации, с чем связано осложненное течение хондродисплазий. Нередко заболевания сопровождаются пороками развития сердца: наличием дополнительных хорд, патологиями клапанного аппарата, поражением крупных эластических сосудов. При вовлечении в процесс мышечной ткани сердца встречаются тяжелые формы кардиомиопатий.
Первичное обследование больных находится в компетенции ортопеда-травматолога, при необходимости к осмотру привлекают генетика, хирурга-ортопеда. Заподозрить наличие хондродисплазии удается по задержке роста в сочетании с множественными скелетными деформациями, однако выделить конкретную нозологическую единицу по физикальным данным затруднительно. План диагностики включает следующие методы исследования:
- Рентгенография костей. Рентгенологические признаки разнообразны: уплощение эпифизарных отделов трубчатых костей, деформация коротких трубчатых костей, неравномерные очаги окостенения. Патогномоничная картина наблюдается при ахондроплазии —на снимке поперечники трубчатых костей увеличены, корковое вещество утолщено, метафизы расширены, рельеф усилен.
- КТ костей скелета. Компьютерная томография имеет более высокую разрешающую способность, поэтому метод рекомендован при затруднениях в дифференциальной диагностике хондродисплазий. Также КТ информативна при мягких фенотипических вариантах, когда отсутствуют выраженные аномалии костного аппарата.
- Генетическое тестирование. На современном этапе медицины расшифрованы гены, отвечающие за большинство вариантов хондродисплазий. Для верификации диагноза используется секвенирование экзона, которое выполняется на базе специализированных генетических центров. По показаниям тестирование проводится ближайшим родственникам больного.
Лечение хондродисплазий
Специфическая терапия не разработана. Суть медицинской помощи заключается в максимально возможном восстановлении нормальных пропорций скелета, улучшении функциональности конечностей, предупреждения тяжелой инвалидности. Важную роль играет разъяснение пациентам недопустимости повышенных нагрузок на суставы, что может стать причиной резкого ухудшения состояния. Комплекс лечебных мероприятий при хондродисплазиях состоит из следующих направлений:
- Ортопедическая коррекция. Использование специальных ортезов, фиксаторов (например, при вывихе тазобедренного сустава) частично компенсирует имеющиеся анатомические аномалии. Чтобы улучшить походку, подбирается специальная ортопедическая обувь.
- Хирургические вмешательства. Хороший функциональный и косметический результат дают операции по удлинению конечностей, которые выполняются в несколько этапов. Также оперативная помощь показана для устранения крупных экзостозов, ликвидации суставных контрактур, коррекции стеноза спинномозгового канала.
- Реабилитация. Больным с хондродисплазиями назначаются индивидуальные курсы лечебной физкультуры, механотерапии, специального массажа. Для уменьшения мышечного напряжения полезны физиотерапевтические процедуры.
Среди хондродисплазий есть летальные варианты, приводящие к гибели плода во внутриутробном периоде или ранней смерти ребенка, и мягкие формы, при которых возможна успешная реабилитация и социализация пациентов. Большое значение для улучшения прогноза имеет своевременная помощь хирургов-ортопедов, всесторонняя реабилитация. Учитывая генетические предпосылки заболевания, эффективные меры профилактики не разработаны.
2. Множественная эпифизарная хондродисплазия: особенности первичного эндопротезирования тазобедренного сустава// А.Ю. Милюков// Политравма. — 2017. — №3.
4. Экзостозная хондродисплазия взрослых (клиника, диагностика, хирургическое лечение)/ А.В. Балберкин. — 1994.
Акрофациальный дизостоз Нагера (acrofacial dysostosis Nager)
Впервые синдром описали в 1948 г. F. Nager и J. de Reynier.
Минимальные диагностические признаки: гипоплазия нижней челюсти, антимонголоидный разрез глаз, стеноз или атрезия слухового канала, гипо- или аплазия I пальца кисти и лучевой кости.
Клиническая характеристика
Для данного синдрома характерно сочетание признаков челюстно-лицевого дизостоза (синдром Франческетти) с недоразвитием I пальца кисти и лучевых костей. Аномалии челюстно-лицевой области включают антимонголоидный разрез глаз, недоразвитие ресниц и колобому нижнего века, резкую гипоплазию нижней челюсти, укорочение твердого неба, гипоплазию зачатков коренных зубов.
Постоянным признаком является снижение слуха вследствие стеноза или атрезии слухового канала. В некоторых случаях обнаруживаются преаурикулярные выросты, деформация и низкое расположение ушных раковин. Поражение конечностей включает укорочение дистальных отделов предплечий, гипо - или аплазию лучевой кости, части костей запястья, пястья, I пальца.
При отсутствии I пальца II палец противопоставлен остальным и может иметь удвоенную дистальную фалангу. Иногда отмечаются искривление V пальца, ограничение подвижности в локтевом суставе, отсутствие II пальца. Больные дети обычно отстают в психомоторном развитии. Популяционная частота неизвестна. Соотношение полов неизвестно.
Тип наследования точно не установлен, возможно, аутосомно-доминантное и аутосомно-рецессивное наследование.
Дифференциальный диагноз: нижнечелюстно-лицевой дизостоз, окуло-аурикуло-вертебральный синдром, окуло-мандибуло-фасциальный синдром.
«Наследственные синдромы и медико-генетическое консультирование»,
С.И. Козлов, Е.С. Еманова
Читайте далее:
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма

Содержит актуальную клиническую информацию по ультрасонографии и ориентирован на врачей ультразвуковой диагностики, выходит с 1996 года.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) - это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза-Франческетти, синдром Франческетти-Цвалена-Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер - имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза-Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
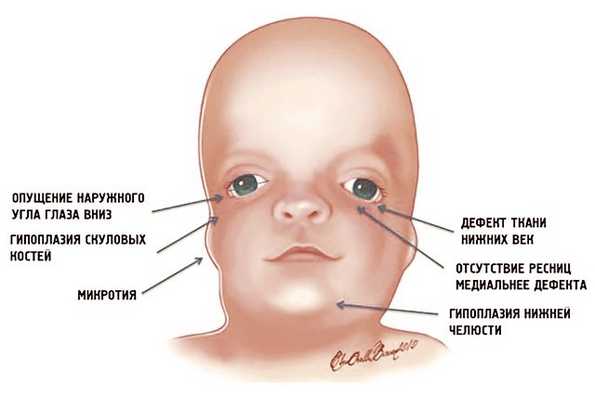
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса 5, также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости 4. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев - это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 - q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4-6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип - мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши - все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности - редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким - 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12-13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2-4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Современные подходы к изучению плодового фенотипа в эру генетического ультразвука
Медико-генетическое отделение Московского областного НИИ акушерства и гинекологии, Москва.
Курс пренатальной диагностики, ФГБОУ ДПО РМАПО Минздрава России, Москва.

УЗИ аппарат RS85
Революционные изменения в экспертной диагностике. Безупречное качество изображения, молниеносная скорость работы, новое поколение технологий визуализации и количественного анализа данных УЗ-сканирования.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика - современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Использование эхографии делает возможным пренатальное выявление не менее 80% врожденных дефектов во II, III триместрах беременности и свыше 50% значимых нарушений анатомии плода уже в I триместре начиная со сроков 11-12 нед. Весь комплекс диагностированных пороков развития и различных микроаномалий в совокупности составляет фенотипический спектр синдрома, который состоит из «ядерных» (главных, ключевых) признаков и дополнительных аномалий, наличие которых не является обязательным для диагностики клинической нозологии синдрома.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1-4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11-14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7-9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).
Читайте также: