Дисплазии черепа с поражением нервной системы
Добавил пользователь Alex Обновлено: 22.01.2026
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Ваша заявка принята!
Благодарим за обращение.
В ближайшее время с вами свяжется наш специалист.
Врожденные пороки, аномалии, дефекты развития головного мозга
Многочисленные пороки развития у детей возникают из-за нарушения внутриутробного эмбриогенеза. Врожденные аномалии у плода и новорожденного ребенка выявляются сразу по внешнему виду черепа или через несколько лет после появления неврологических расстройств.
Миграционные нарушения церебральных тканей в зависимости от локализации обуславливают определенные клинические симптомы. Острое течение патологии позволит неврологам установить диагноз своевременно. Хроническое развитие с циклами обострений и ремиссий не является специфическим признаком нозологии.
Дисплазия головного мозга - что это такое
Ограниченное церебральное повреждение на ограниченном участке провоцирует разнообразные клинические проявления. Эпилептические приступы сочетаются с нарушением сознания, кортикальными нарушениями.
Первоначальные признаки нозологии с помощью электроэнцефалографии. Небольшие церебральные изменения устраняются антиконвульсивными препаратами.
Разновидности фокальной дисплазии:
- Первый тип - изменение корковой архитектоники локальное;
- Второй тип - очаговые цитоархитектонические изменения;
- Третий тип - патология архитектоники при вторичных болезнях (темпоральный склероз, церебральная мальформация, энцефалит Расмуссена, ишемия).
Способы лучевой нейровизуализации МРТ головного мозга в Санкт-Петербурге помогают верифицировать стадию нозологии.
Поликистозная дисплазия мозга характеризуется наличием множества кистозных разрастаний внутри церебральной ткани.
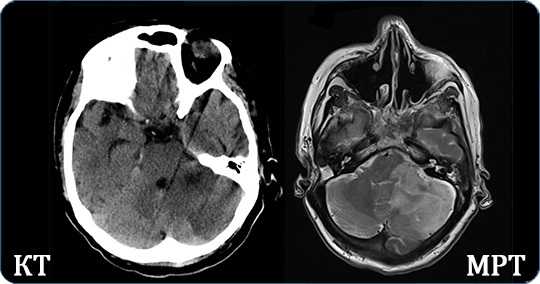
Для диагностики пороков сравнивают результаты КТ и МРТ
Аномалии развития ствола головного мозга - причины возникновения
В зависимости от морфологических изменений выделяют группы аномалий мозга:
- Положения;
- Количества;
- Размера и формы;
- Структуры (строения).
Первая группа нозологий возникает по причине недоразвитие зачатка церебральной структуры или полного отсутствия эмбрионального зачатка. Если ребенок рождается нормальным, прогноз является неблагоприятным из-за отсутствия части мозга.
К аномалиям положения следует отнести удвоение органа, слияние между собой нескольких частей одновременно.
Дефекты положения церебральных структур
Все нозологические формы группы определяются тремя факторами:
- Инверсионное смещение органа относительно собственной оси, срединного положения;
- Дистопия - необычная локализация эмбриональных структур;
- Гетеротопия - патология закладки органа.
Степень смещения определяется выраженность клинических симптомов, длительность жизни человека.
Пороки размеров и формы мозга
Перечень нозологий данной категории определяется рядом морфологических изменений:
- Синостозы нескольких органов;
- Гиперплазия — увеличение количества тканей и размеров церебральных тканей;
- Гипоплазия диспластическая - уменьшение размеров структуры;
- Гипоплазия простая - нет изменений морфологии.
Дефекты строения головного мозга
Нозология сопровождается аномалиями естественного образования отверстия, морфологических особенностей структуры. Гетероплазия развивается на этапе внутриутробного развития. Характеризуется атипичным формированием ткани.
Дисплазия - патология соотношения суставных поверхностей.
Гамартрия - неправильное развитие тканевых структур. Стенотическое сужение канала, протока - бывает врожденным и приобретенным.
Дизонтогенетическая киста сопровождается значительным сужением компенсаторных возможностей органа.
Классификация пороков эмбрионального развития:
- Фетопатии;
- Эмбриопатии;
- Бластопатии;
- Гаметопатии.
В зависимости от времени появления дефектов эмбрионального развития возникает определенный тип нарушений.
По обширности повреждения выделяют виды церебральных дефектов:
- Множественные - поражают сразу несколько мозговых областей;
- Системные - локализуются в пределах одного участка;
- Изолированные - обеспечивают повреждение одного органа.
Врожденные аномалии центральной нервной системы могут провоцироваться инфекционными агентами:
- Токсоплазма;
- Цитомегаловирус;
- Вирус Коксаки.
Встречаются алкогольные аномалии, если беременная женщина употребляла спирт во время вынашивания плода. Патология провоцируется хромосомными аберрациями, генетическими мутациями во время формирования нервной трубки (третья-четвертая неделя беременности).
Основные виды мозговых дефектов
Выделяют дефекты формы, размеров, расположения отдельных анатомических структур. Рассмотрим основные виды врожденных аномалий развития головного мозга.
Что такое энцефалоцеле
Проникновение церебральных тканей через дефекты черепа обуславливает разные неврологические симптомы, зависящие от особенностей участка выпадения. Небольшая анэнцефалия напоминает кефалогематому, но рентгенография черепа выявляет незаращение по средней линии, ассиметричные участки.
С помощью хирургического вмешательства удается устранить патологию, но крупные очаги нельзя устранить эктопированием выпячивания. Энцефалоцеле верифицируется методами лучевой нейровизуализации - МРТ и КТ.
Особенности анэнцефалии
Патология характеризуется отсутствием отдельных костей черепа. Места разрушения зарастают соединительной тканью, что не позволяет оптимально регулировать внутричерепное давление.
Большая часть нозологических форм не совместима с жизнью. Смертельный исход возникает сразу после рождения, когда раскрываются легкие, начинается подача кислорода к церебральной паренхиме.
Проявления микроцефалии
Недоразвитие церебральных тканей формируется у одного ребенка на пять тысяч новорожденных детей. Определяется нозология по уменьшению размера черепной коробки, нарушением соотношения между мозговой и лицевой частью черепа.
Микроцефалия (синдром Джакомини) развивается внутриутробно у женщин с инфекциями, паразитарными болезнями.
Причины первичной микроцефалии:
- Генетические аномалии с передачей по аутосомно-рецессивному типу;
- Токсоплазмоз, энцефалит цитомегаловирус, краснуха.
Этиологические факторы вторичной микроцефалии:
- Церебральные кисты;
- Обызвествления и кровоизлияния внутрь мозговой паренхимы.
Уменьшением размеров черепа характеризуется около десяти процентов олигофрений. С ранних лет у ребенка прослеживается отставание развития, закрытые пороки развития. Умственная отсталость сопровождается судорожным синдромом. Мышечные подергивания сопровождаются неправильным развитием мозговой части черепной коробки.
Чем характеризуется макроцефалия
Диагностировать патологию позволяет увеличение черепной коробки. Нозология характеризуется гипертрофией одного полушария, непропорциональным развитием одной половины. Умственное недоразвитие - самое частое проявление. Судорожные припадки прослеживаются примерно у девяти-десяти процентов пациентов.
Клиника нозологии появляется в течение первых лет жизни, что позволяет своевременно верифицировать патологию. Мозговые миграционные нарушения слишком серьезны для эффективного лечения болезни.
Симптоматика голопрозэнцефалии
Голопрозэнцефалия - болезнь сопровождается дефектом развития полушарий. Отсутствие разделения между церебральными половинами обуславливает изменения активности функциональных центров.
Сильные дисплазии приводят к аномалиям желудочков, асимметричностью лицевой и мозговой части. Выраженные дефекты приводят к омертвению церебральной паренхимы с высокой вероятностью гибели в первые сутки после появления ребенка на свет.
Образование единой полусферы является врожденным пороком из-за генетической аномалии тринадцатой-пятнадцатой хромосом. Нозология нередко сочетается с другими пороками развития:
- Циклопия;
- Этмоцефалия;
- Цебоцефалия.
Заболевание сопровождается мертворождением. Способность к выживанию минимальна. Прогноз неблагоприятный.
Клиника дисплазии церебральной кистозной
Множественные кистозные полости обуславливают миграционные нарушения. Дефекты развития сопровождаются аномалиями распределения спинномозговой жидкости. Многочисленные кисты нельзя удалить. Нередко провоцируют мышечные судороги. Низкая эффективность антиконвульсивного лечения приводит к прогрессированию симптомов.
Единичные кисты могут не представлять опасности. Возможно субклиническое протекание симптоматики на фоне увеличения внутричерепной гипертензии.
Чем проявляется агирия
Лиссэнцефалия - это дефекты формирования архитектоники мозговой коры. Тяжелые судороги обуславливает недоразвитие церебральных извилин. Нозология формирует моторные и психические проявления. Неврологические признаки заболевания - синдром Леннокса-Гасто, Веста.
Слаженность головного мозга провоцирует параличи, парезы, полиморфные судороги. Признаки нозологии развиваются на первом году жизни. Примерно данный промежуток времени живет малыш.
Клинические признаки пахигирии
Дефект развития обусловлен отсутствием формирования вторичных и третичных извилин. Выпрямление борозд второго типа нарушает церебральную архитектонику.
Патология послойного строения коры характеризуется гетеротопией нервных клеток. Хорошо показывает пахигирию МРТ.
Клинические симптомы краниостеноза
Болезнь характеризуется сужением черепа с компрессией головного мозга между костями. В зависимости от прогрессирования выделяют декомпенсированный и компенсированный вариант нозологии.
По особенностям течения выделяют стабильную и прогрессирующую формы болезни. Чаще причины обусловлены ранним зарастанием коронарного или сагиттального швов. Патология без своевременного лечения приводит к летальному исходу, так как появляется ущемление головного мозга. Клинические симптомы зависят от преимущественной локализации зоны сдавления белого вещества и паренхимы.
Неврологическая симптоматика характеризуется многочисленными расстройствами на фоне увеличения внутричерепного давления.
У ребенка с краниостенозом сильная головная боль, поэтому малыш беспокоен, раздражителен, плаксив. Потеря памяти, нарушение концентрации внимания возникает при длительном сохранении состояния. Прогноз патологии неблагоприятный.
Показатели агенезии мозолистого тела
Нозология характеризуется недоразвитием мозолистого тела. Сопутствующая патология - недоразвитие третьего желудочка мозга. Гипоплазия провоцирует недоразвитие мышечной мускулатуры, парезы и параличи, мышечные судороги.
Проявления синдрома Айкарди возникают при сочетании недоразвития мозолистого тела с хориоретинальными пороками. Клиническая картина дополняется миоклоническими судорогами, формированием многочисленных лакунарных узлов в сетчатке глаза, диске зрительного нерва. Нозология характеризуется микрофтальмом, маятникообразными движениями глаз.
Некоторые исследователи выявляют у пациентов с агенезией мозолистого тела дефекты Х-хромосомы у мужчин.
Клиника микрополигирии
Заболевание возникает по причине формирование многочисленных мелких извилин. Нормальная церебральная кора имеет шесть слоев. При аномалии прослеживается четыре слоя. Аномальная структура приводит к клиническим симптомам:
- Расстройство глотания;
- Патология жевательных, мимических мышц;
- Олигофрения;
- Параплегия лица.
Дебют болезни наблюдается на первом году жизни.
Клинические проявления гетеротопии
Нозология возникает из-за нейрональной миграции. Отсутствие передачи нервного сигнала возникает из-за гетеротопионов - патологические скопления в виде ленточной или узловой формы.
Из-за гетеротопии появляется олигофрения, эпилептический синдром, разные мышечные судороги.
Диагностика врожденных пороков головного мозга
Большинство нозологических форм обнаруживается вначале по клиническим проявлениям. Легкое течение, гипотонические сокращения мускулатуры провоцируют судорожный синдром у детей первого года жизни.
Исключить гипоксические и травматологические проявления позволяют инструментальные способы диагностики - УЗИ, нейросонография, МРТ и КТ. Процедур достаточно для выявления аномалий развития, кист, гетеротопических участков.
Электроэнцефалография обнаруживает зоны повышенной церебральной активности при присутствии судорожных подергиваний мышц. Врожденные виды требуют генетической диагностики для исследования ДНК, определения мутаций хромосомного аппарата.
Краниометафизарная дисплазия
Краниометафизарная дисплазия - наследственное состояние из группы остеохондродисплазий, характеризующееся аномалиями развития черепа и метафизов костей конечностей. Симптомами данного заболевания являются гипертелоризм, пороки развития лица, нередко уродующие больного, аномальное формирование носовых ходов с нарушением их проходимости, иногда головные боли и искривления конечностей. Диагностика краниометафизарной дисплазии осуществляется на основании изучения настоящего статуса пациента, рентгенологических данных и результатов молекулярно-генетических исследований. Специфического лечения данной патологии не существует, используют симптоматическую терапию, в некоторых случаях проводят хирургические вмешательства для облегчения дыхания пациента и улучшения его внешнего вида.
Общие сведения
Краниометафизарная дисплазия - группа наследственных заболеваний, которые приводят к порокам развития костей лицевого и мозгового отделов черепа различной степени выраженности в сочетании с аномалией метафизов длинных трубчатых костей и другими нарушениями. Ранее это состояние относили в одну группу с болезнью Пайла (множественная метафизарная дисплазия, краниометафизарная дисплазия Пайла), однако в настоящее время его выделяют в отдельную нозологическую единицу. Это связано с тем, что при данном заболевании превалирующими являются именно аномалии строения черепа (деформации, гиперостозы и склерозы костей), тогда как патологии иных отделов скелета выражены слабо. Существуют две основные формы краниометафизарной дисплазии, отличающиеся между собой механизмом наследования (аутосомно-доминантный и аутосомно-рецессивный типы), клиническими проявлениями и выраженностью нарушений. За счет наследования посредством аутосом патология с равной долей вероятности поражает как мужчин, так и женщин. Встречаемость краниометафизарной дисплазии точно не установлена, доминантный тип регистрируется во много раз чаще рецессивного.
Причины краниометафизарной дисплазии
Основной причиной более частой, но более легкой аутосомно-доминантной формы краниометафизарной дисплазии становятся мутации в гене ANKH, располагающемся на 5-й хромосоме. Ген кодирует белок, который является мембранным переносчиком пирофосфата, участвующего в угнетении процессов обызвествления костной ткани и ее резорбции. В результате генетического дефекта у белка-переносчика изменяется структура, и он становится неспособным полноценно выполнять свои функции, что приводит к краниометафизарной дисплазии. На клеточном уровне это проявляется аномальным изменением активности остеокластов, развитием склероза и гиперостоза главным образом костей черепа. Также может нарушаться конфигурация основных отверстий основания черепа, что становится причиной компрессии некоторых сосудов и нервов и во многом определяет остальные симптомы краниометафизарной дисплазии (нарушения слуха, головные боли, поражения тройничного и лицевого нервов). В генетике известны и другие заболевания, обусловленные поражением гена ANKH, в частности - наследственная псевдоподагра или семейный хондрокальциноз.
Намного более редкая аутосомно-рецессивная форма краниометафизарной дисплазии обусловлена дефектом гена GJA1, локализованного на 7-й хромосоме. Продуктом его экспрессии является белок под названием коннексин 43, принимающий активное участие в формировании межклеточных (межщелевых) контактов во многих тканях, что позволяет клеткам обмениваться низкомолекулярными соединениями. Патогенез развития краниометафизарной дисплазии при миссенс-мутации c716G>A неизвестен, основная проблема сводится к выявлению причин изолированного поражения костей черепа и метафизов костей при относительном отсутствии патологий других органов. Учитывая, что наибольшее количество коннексина 43 у человека находится в сердечной ткани, вопрос отсутствия патологий сердца при данной мутации на сегодняшний день представляет собой загадку для большинства врачей-генетиков. Так же, как и в случае аутосомно-доминантной формы краниометафизарной дисплазии, при этом типе заболевания наблюдаются многочисленные вторичные нарушения, обусловленные воздействием измененных костей и отверстий на нервные структуры.
Симптомы краниометафизарной дисплазии
Аутосомно-доминантный тип краниометафизарной дисплазии характеризуется более легким течением, нередко при рождении ребенка никаких симптомов заболевания не выявляется. Лишь к первому году жизни возникает гипертелоризм с расширением переносицы вследствие гиперостоза носовых костей. В дальнейшем краниометафизарная дисплазия приводит к сужению носовых ходов и нарушению их проходимости, поэтому у больных нередко приоткрыт рот из-за нарушенного носового дыхания. К 6-7 годам может начать определяться увеличение метафизов длинных трубчатых костей, что внешне проявляется увеличением размеров коленных и локтевых суставов. Примерно у половины больных краниометафизарной дисплазией развиваются нарушения слуха различной выраженности вплоть до полной глухоты - чаще всего это обусловлено компрессией слухового нерва в костном канале. Из-за сдавления нервов возникают и другие неврологические нарушения, возможны расстройства чувствительности на лице, невралгия тройничного нерва и головные боли.
Краниометафизарная дисплазия аутосомно-рецессивного типа характеризуется намного более выраженными пороками развития костей черепа и конечностей, признаки патологии часто выявляются сразу после рождения ребенка. У больных присутствует выраженный гипертелоризм, черты лица зачастую крайне несимметричны, деформации переносицы и носа могут приобретать признаки уродства. В ряде случаев наблюдаются макроцефалия, нижнечелюстной прогнатизм, другие нарушения прикуса и расположения зубов. По мере роста больного аномалии костей черепа могут усугубляться. Метафизы костей конечностей резко расширены, что нередко обуславливает вторичные деформации (например, Х-образное искривление ног). Как и в предыдущем варианте, при этой форме краниометафизарной дисплазии часто возникают разнообразные неврологические нарушения, вызванные сдавлением и травматизацией черепно-мозговых нервов. Они могут проявляться глухотой, нарушениями зрения, расстройствами кожной чувствительности на лице, парезом мимической мускулатуры и головными болями. Существуют отдельные описания больных, одновременно страдающих краниометафизарной дисплазией и умственной отсталостью, однако достоверных данных о взаимосвязи этих двух состояний на сегодняшний день нет.
Диагностика и лечение краниометафизарной дисплазии
Диагностика краниометафизарной дисплазии основывается на данных осмотра пациента, изучении его наследственного анамнеза, результатах рентгенологических исследований и молекулярно-генетических анализов. При осмотре определяются различные по выраженности аномалии развития костей черепа, что отражается на чертах лица больного. При этом аутосомно-доминантная форма, особенно у маленьких детей, довольно слабо проявляет себя в отношении этого симптома. При обоих типах краниометафизарной дисплазии практически всегда присутствуют гипертелоризм, расширение переносицы за счет разрастания носовых костей по направлению к скулам, сужение или непроходимость носовых ходов. В рамках физикального осмотра и дополнительных исследований могут быть также выявлены неврологические нарушения: снижение или отсутствие слуха, снижение зрения, симптомы со стороны тройничного и лицевого нервов.
Намного больше информации при краниометафизарной дисплазии дают рентгенологические методы исследования скелета. На рентгенограммах черепа при аутосомно-доминантной форме заболевания определяются уплотнение костной ткани в области затылочной кости, склероз основания черепа, пониженная пневматизация синусов и ячеек височной кости. В ряде случаев может выявляться склероз межкостных швов и расширение метафизов трубчатых костей. При аутосомно-рецессивном типе краниометафизарной дисплазии на рентгенограммах обнаруживаются схожие, но намного более выраженные нарушения, например - полное отсутствие околоносовых пазух, резкое сужение и иногда заполнение костной тканью отверстий черепно-мозговых нервов. Кроме того, наблюдается склероз не только основания, но и свода черепа, в ряде случаев определяются значительные деформации костей лицевого отдела. Немного сильнее, чем при доминантном типе, выражены расширение и склероз метафизов трубчатых костей.
Изучение наследственного анамнеза и генетическая диагностика также активно используются для определения краниометафизарной дисплазии. При этом можно сначала установить тип наследования заболевания, что позволяет скорректировать молекулярно-генетический анализ для поиска мутаций в конкретном гене. Для этого применяют метод прямого секвенирования генов ANKH и GJA1.
Специфического лечения краниометафизарной дисплазии не существует, назначают симптоматическую терапию с привлечением разнообразных хирургических техник. Последние, в частности, могут улучшить проходимость носовых ходов для облегчения дыхания. Уменьшить выраженность неврологической симптоматики можно путем расширения отверстий соответствующих черепно-мозговых нервов. Кроме того, пластические хирурги способны минимизировать выраженность эстетических дефектов при краниометафизарной дисплазии. Для улучшения слуха используют слуховые аппараты.
Прогноз и профилактика краниометафизарной дисплазии
Во многих случаях прогноз краниометафизарной дисплазии аутосомно-доминантного типа относительно выживаемости больного благоприятный - ни аномалии черепа, ни вторичные неврологические нарушения не приводят к тяжелым последствиям. Возникающие в детстве патологии слуха или зрения могут медленно прогрессировать до 20-30 лет, после этого ухудшений обычно не происходит. Аутосомно-рецессивный тип краниометафизарной дисплазии характеризуется более неблагоприятным прогнозом, поскольку костные нарушения черепа при этом состоянии имеют тенденцию к усугублению и могут с каждым годом все сильнее травмировать нервы. В некоторых случаях это приводит к параличам и расстройствам со стороны вегетативной нервной системы. Профилактика краниометафизарной дисплазии не разработана, для уменьшения риска развития осложнений при наличии такого состояния необходимо проводить регулярное обследование у невролога.
Фокальная корковая дисплазия
Фокальная корковая дисплазия — аномалия структуры коры головного мозга, затрагивающая её ограниченный участок. Клинически проявляется фокальными двигательными эпиприступами с потерей сознания, но небольшой продолжительностью. Диагностируется неврологом или эпилептологом по данным ЭЭГ, специально проведённого МР-сканирования, субдуральной электрокортикографии, ПЭТ головного мозга. Как правило, эпилепсия при корковой дисплазии устойчива к проводимой противоэпилептической терапии. Альтернативным методом лечения выступает нейрохирургическая резекция участка дисплазии.
Фокальная корковая дисплазия (ФКД) — возникшее в период внутриутробного развития локальное нарушение в строении мозговой коры. Является самым частым этиофактором развития эпилепсии у детей. По данным международного исследования ILAE (2005 г.) ФКД была диагностирована у 31% детей с эпилепсией. Отличительными особенностями эпилептических пароксизмов при ФКД являются: устойчивость к проводимой противоэпилептической терапии, агрессивное течение с развитием у детей задержки психического развития и эпилептической энцефалопатии, эффективность нейрохирургических способов лечения.
Локальные диспластические изменения коры располагаются преимущественно в височных и лобных долях. Они слабо заметны на макроскопическом уровне, что затрудняет диагностику ФКД даже при помощи таких современных методов нейровизуализации как МРТ. Проблемы диагностики ФКД особо актуальны в практической неврологии и педиатрии, поскольку её выявление как причины эпилептических пароксизмов имеет решающее значение для выбора эффективной лечебной тактики в отношении резистентных форм эпилепсии.
Причины фокальной корковой дисплазии
ФКД обусловлена нарушением окончательных этапов развития церебральной коры (кортикогенеза) во внутриутробном периоде. В результате нарушений миграции и дифференцировки клеток коры образуется участок с аномальными нейронами, патологической утолщенностью, уплощенностью извилин или изменённой архитектоникой (появлением клеток, типичных для одного слоя, в другом слое коры). Формирование фокальной корковой дисплазии происходит незадолго до родов — за 4-6 недель до окончания периода внутриутробного развития. Более тяжёлые формы пороков мозговой коры (например, полимикрогирия, гемимегалэнцефалия) связаны с нарушениями конца 2-го начала 3-его триместра беременности.
Нельзя исключить генетическую природу ведущего к ФКД сбоя в кортикогенезе. Так, у многих пациентов обнаружены изменения в гене TSC1, которые также наблюдаются при туберозном склерозе. В настоящее время проводятся международные исследования по поиску генетического субстрата корковых дисплазий.
Классификация фокальной корковой дисплазии
До недавнего времени выделяли 2 основных типа ФКД. В 2011 г. была разработана новая классификация, в которую включён 3-й тип, ассоциированный с другим основным поражением церебральных структур. Согласно этой классификации выделяют:
ФКД I типа — локальное нарушение архитектоники коры: радиальное (IA), тангенциальное (IB) или смешанное (IC). Выявляется у 1,7% обследованных практически здоровых людей.
ФКД II типа — очаговое нарушение цитоархитектоники с наличием аномальных нейронов (IIA) и так называемых баллонных клеток (IIB). Как правило, дисплазия затрагивает лобные доли.
ФКД III типа — вторичное нарушение корковой архитектоники, обусловленное другой патологией: мезиальным темпоральным склерозом (IIIA), глиальной опухолью (IIIB), мальформацией сосудов головного мозга (IIIC) или другими нарушениями (IIID) — энцефалитом Расмуссена, нейроинфекцией, посттравматическими или постишемическими изменениями и пр.
Симптомы фокальной корковой дисплазии
Ведущим клиническим проявлением ФКД выступает фокальная эпилепсия. Как правило, она манифестирует в детском возрасте. Эпилептические пароксизмы отличаются своей кратковременностью — длятся не более минуты. Среди них преобладают сложные (с расстройством сознания) фокальные моторные приступы, зачастую с автоматизмами в начальном периоде пароксизма. Спутанность сознания в постприступный период выражена незначительно. Характерны двигательные феномены и внезапные падения. Вторичная генерализация эпиприступов происходит заметно быстрее, чем при височной эпилепсии.
Возраст дебюта эпилепсии и сопутствующая клиническая симптоматика зависят от типа, выраженности и расположения очага корковой дисплазии. Ранняя манифестация аномалии обычно сопровождается задержкой психического развития ребёнка и когнитивными нарушениями.
ФКД I типа имеет менее тяжёлое течение и не всегда проявляется эпиприступами. У ряда пациентов она приводит к затруднению в познавательной деятельности и проблемам в обучении. ФКД II типа сопровождается тяжёлыми парциальными и вторично генерализованными эпиприступами. У многих пациентов наблюдается эпилептический статус. Клиника и течение ФКД III типа зависит от характера основной патологии.
Диагностика фокальной корковой дисплазии
Основной метод диагностики ФКД — магнитно-резонансная томография. Она должна выполняться по специальному протоколу с толщиной срезов 1-2 мм. Только такое тщательное сканирование способно выявить минимальные структурные изменения мозговой коры. В МРТ диагностике корковой дисплазии имеет значение опыт и квалификация рентгенолога. Поэтому при необходимости результаты исследования следует показать более опытному в этом вопросе специалисту.
К МРТ признакам ФКД относятся: локальная гипоплазия или утолщение коры, «смазывание» перехода между белым и серым веществом, изменённый ход извилин, повышенный МР-сигнал на ограниченном участке коры при исследовании в режимах Т2 и FLAIR. Каждый тип ФКД имеет свои особенности МРТ-картины.
В обязательно порядке пациентам с ФКД проводится электроэнцефалография. В большинстве случаев она выявляет очаговую эпилептическую активность мозга не только в момент приступа, но и в межприступный период. Во время приступа отмечается повышенная возбудимость и активация зон коры, прилежащих в визуализируемому на МРТ очагу дисплазии. Это связано с наличием аномальных клеток и за пределами основного участка корковой дисплазии, который является лишь «верхушкой айсберга».
Выявление зоны начала эпилептического приступа возможно при помощи ПЭТ, совмещённой с МРТ-изображением. При этом радиофармпрепарат должен быть введён пациенту после первого же пароксизмального разряда. Такое исследование особенно ценно при МРТ-негативных случаях ФКД и при несовпадении очага, визуализируемого на МРТ, с данными ЭЭГ. Для более точного определения расположения эпилептогенного очага проводится инвазивная электрокортикография с установкой субдуральных электродов, требующая краниотомии.
Лечение фокальной корковой дисплазии
Терапию начинают с подбора эффективного противосудорожного препарата и его дозы. Пациента совместно курируют эпилептолог и невролог. Возможно применение карбамазепина, препаратов вальпроевой к-ты, диазепама, леветирацетама, топирамата и др. антиконвульсантов. Однако зачастую эпилепсия при ФКД оказывается резистентной к противосудорожной терапии. В таких случаях ставится вопрос о хирургическом лечении и проводится консультация нейрохирурга.
Поскольку диспластические изменения носят фокальный характер, то хирургическое удаление патологического очага является эффективным способом лечения ФКД. В начале нейрохирургического вмешательства проводится электростимуляция и индивидуальная интраоперационная кортикография с составлением карты функционально важных участков коры, что позволяет избежать их травмирования в ходе операции. Многие нейрохирурги настаивают на целесообразности как можно более радикального удаления диспластического очага для достижения наилучших результатов лечения. Сложность заключается в широком распространении зоны точечно расположенных патологически изменённых клеток вокруг основного очага и невозможности их полного удаления. Распространённые и билатеральные эпилептогенные поражения являются противопоказанием к хирургическому лечению.
В зависимости от локализации и распространённости очага применяется один из 3 видов оперативных вмешательств: селективная резекция эпилептогенной зоны, стандартизированная резекция головного мозга (лобэктомия), тэйлорированная резекция — «выкраивание» зоны дисплазии, определённой в ходе кортикографии. При ФКД III типа зачастую требуется удаление и дисплазии, и основного очага поражения (опухоли, участка склероза, сосудистой мальформации и т. п.).
Прогноз при фокальной корковой дисплазии
Прогноз зависит от типа ФКД, своевременности проведённого лечения, радикальности удаления участка корковой дисплазии. Консервативная терапия, как правило, не даёт желаемого результата. Длительное течение эпилепсии в детском возрасте чревато нарушением нервно-психического развития с исходом в олигофрению.
Хирургическое лечение наиболее эффективно при единичном хорошо локализуемом очаге. По некоторым данным полное отсутствие пароксизмов или их значительное урежение наблюдается у 60% прооперированных пациентов. Однако спустя 10 лет приступы отсутствуют только у 32%. По всей видимости, рецидив эпилепсии в таких случаях связан с неполным удалением эпилептогенных элементов.
Стойкие послеоперационные неврологические расстройства отмечаются в 2% случаев, при распространённых поражениях — в 6%. Риск их развития повышен при проведении лобэктомии и вмешательствах вблизи функционально значимых участков коры.
Аномалии развития головного мозга ( Пороки развития головного мозга )
Аномалии развития головного мозга — это результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
МКБ-10
Причины
Наиболее весомой причиной сбоев внутриутробного развития является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев. Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ.
Различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
Патогенез
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества.
Виды аномалий мозга
Анэнцефалия — отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле — пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия — уменьшение объема и массы головного мозга, обусловленное задержкой его развития. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия — увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия — характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия — отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия (гладкий мозг, лиссэнцефалия) — отставание развития извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия — укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия — поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия (ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии — скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Диагностика
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Лечение аномалий мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз и профилактика
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии. Эффективной мерой профилактики служит исключение эмбриотоксических и тератогенных влияний на женщину в период беременности. При планировании беременности будущим родителям следует избавиться от вредных привычек, пройти генетическое консультирование, обследование на наличие хронических инфекций.
Дисплазия соединительной ткани у детей и взрослых
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
О таком синдроме, как дисплазия соединительной ткани, говорят тогда, когда организм человека с рождения склонен к нарушениям формирования хрящевой ткани суставов, а также прочих тканей. Малыша, страдающего дисплазией, выявить относительно легко: он отличается необычной гибкостью, его суставы без проблем прогибаются в разные стороны.
У пациентов с дисплазией соединительной ткани уже в молодом возрасте развивается ранний остеохондроз, отмечаются нарушения зрения, пороки клапанов сердца. Соответственно, такие люди быстро обретают разные проблемы со здоровьем - в частности, с опорно-двигательным аппаратом.
Код по МКБ-10
Эпидемиология
О дисплазии соединительной ткани говорят в случаях, когда имеются признаки нарушенного соединительнотканного развития на эмбриональном и постнатальном этапе, и эти нарушения вызывают сбой гомеостаза. Расстройство происходит на уровне тканей, органов и всего организма в целом: отмечаются всевозможные морфофункциональные патологии.
Распространенность и частота заболеваний соединительной ткани, о которых сообщают, весьма различны, в зависимости от различий в методологии исследования. [1] Проблема недифференцированной дисплазии соединительной ткани (нДСТ) является актуальной по причине значительной частоты встречаемости данной патологии в популяции взрослого населения в целом, и в частности, среди женщин репродуктивного возраста (7-8%). [2] За помощью медиков пациенты с дисплазией обращаются в шесть раз чаще, нежели больные другими заболеваниями.
Заболеваемость не связана с половой и расовой принадлежностью пациентов.
Причины дисплазии соединительной ткани
Дисплазия соединительной ткани - это синдром, который включает в себя обширный ряд патологий. Причинами выступают расстройства, связанные с генетическими нарушениями построения коллагеновых соединительнотканных волокон. Процесс преимущественно захватывает костную ткань, связочный и сухожильный аппарат и кожные покровы.
Базовым механизмом соединительнотканных нарушений являются генные мутации. Особую роль играют изменения в генах, ответственных за продукцию основного белкового вещества, составляющего соединительную ткань - речь идет о коллагене (иногда - о фибриллине). Когда в ходе формирования белковых волокон происходят болезненные изменения, то они становятся менее прочными, неспособными выдержать нагрузку. Дополнительным фактором развития синдрома может стать недостача магния в организме.
Факторы риска
Учеными доказано, что развитию дисплазии соединительной ткани у ребенка способствуют такие факторы:
- анемия у матери при беременности;
- угроза прерывания беременности;
- хроническая недостача кислорода у плода;
- хроническая фетоплацентарная недостаточность;
- сильный или продолжительный токсикоз, гестоз;
- сопутствующие беременности хронические патологии (заболевания эндокринной системы, почек, органов желудочно-кишечного тракта или респираторных путей).
Гетерозиготные мутации в гене коллагена типа II (COL2A1) приводят к группе скелетных дисплазий, известных как коллагенопатия типа II (COL2pathy). [3], [4], [5] Цепи proα1 (I) и proα2 (I) коллагена 1 кодируются генами COL1A1 и COL1A2 соответственно; Количественные или качественные дефекты синтеза коллагена I типа обычно проявляются в виде коллагенопатии I типа и несовершенного остеогенеза. Большинство пациентов (около 90%) с клиническим диагнозом несовершенного остеогенеза имеют мутацию в генах COL1A1 или COL1A2 с аутосомно-доминантным типом наследования. Шесть других генов, CRTAP, LEPRE1, FKBP10, PP1B, SP7 / Osterix (OSX) и SERPINH1, связаны с аутосомно-рецессивными формами. [6], [7], [8]
Базовый механизм развития дисплазии соединительной ткани, равно как и недифференцированной формы заболевания, обусловлен генной мутацией, с вовлечением генов, отвечающих за выработку и диссимиляцию строительных белковых компонентов соединительной ткани, либо ферментных веществ, принимающих участие в указанных процессах. Изменяется количественное формирование качественных составляющих экстрацеллюлярного матрикса, расстраивается фибриллогенез. Генетические детерминанты осуществляются в зависимости от внешних факторов, или практически не зависят от них: это отмечается при дисплазии и недифференцированной дисплазии соответственно. Для соединительнотканной дисплазии присуща полигенность и мультифакторность (патология с генетической предрасположенностью): речь идет о мутации сразу многих генов, а случайное перераспределение отцовских и материнских аллелей постоянно влечет за собой образование следующего единственного в своем роде генотипа.
Факторы при рождении - например, витаминная или макро и микроэлементарная недостача - становятся базовыми причинами, создающими предпосылки для развития дисплазии соединительной ткани. Витамины B-группы стабилизируют обмен белков, аскорбиновая кислота с токоферолом потенцируют адекватную выработку коллагена, а также выступают в роли антиоксидантов. Микро и макроэлементы - медь, бор, цинк и кремний, фтор и кальций, марганец и магний, ванадий, фосфор и селен - выступают кофакторами ферментных веществ, стимулирующих выработку коллагена и насыщение костей минералами. Немаловажно и их участие в электролитном обмене и поддержании кислотно-щелочного равновесия. Калиевые, магниевые и цинк-ионы поддерживают костный рост и усиливают минеральную концентрацию ткани кости. В развитии заболевания любой из указанных факторов имеет первостепенное значение. [9]
Симптомы дисплазии соединительной ткани
Первые признаки дисплазии соединительной ткани проявляются ещё в раннем детском возрасте. Это может быть, как чрезмерная гибкость и гиперподвижность, так и ограниченная мобильность суставов по типу контрактур. Случаются также физические дефекты развития (карликовость), связочная слабость, хрупкие костные ткани, различные искривления позвоночника, плоскостопие, деформированная грудная клетка и пр.
Признаки дисплазии отмечаются и по отношению к другим органам: болезнь может поражать сердце, сосудистую сеть, глаза.
Часто страдает позвоночный столб: позвонки смещаются настолько, что при малейшем движении происходит сдавливание сосудов, ущемляются нервные окончания, возникают боли, нарушается сознание. [10]
Клиническая картина заболевания поражает своим разнообразием, и в этом заключается огромный «минус», поскольку идентифицировать патологию становится очень сложно. Поэтому врачи вынуждены прибегать сразу к нескольким методам лабораторной диагностики, а также к инструментальным видам исследования.
Фенотипические признаки при дисплазии соединительной ткани не всегда присутствуют с рождения и могут проявляться на протяжении всего жизненного периода. Со временем, через годы, чаще всего - под воздействием определенных неблагоприятных условий численность диспластических симптомов и их выраженность увеличивается и усиливается, поскольку первичные нарушения гомеостаза нарастают. В данном случае неблагоприятными условиями могут стать неправильное питание, плохая экология, регулярные интеркуррентные патологии, частые стрессы и пр. Первоочередно затрагивается постоянство присутствия микро и макроэлементов, которые непосредственно участвуют в процессах коллагеновой выработки, а также в регуляции ферментной активности, необходимой для быстрого и качественного синтеза.
В общем, указанные процессы преимущественно зависимы от равновесия содержания кальция и магния в организме. Например, недостача магния на фоне нормы или превышения уровня кальция приводит к повышению активности протеолитических ферментных веществ, которые вызывают коллагеновую деградацию. Как следствие - тяжелая клиническая картина дисплазии соединительной ткани.
Магний регулирует утилизацию кальция в организме. При дефиците магния кальций откладывается в костных и мягких тканях разных органов. При избытке магния кальций начинает плохо усваиваться и выводиться из организма.
Длительный недостаток магния может вызывать признаки ангиоспазма, повышения артериального давления, миокардиальной дистрофии, тахикардии, аритмии, повышенного тромбообразования. Возможны психоневрологические расстройства: невнимательность, депрессия, фобии или тревожные состояния, вегетативные нарушения, головные боли и головокружения, бессонница, онемения конечностей. Висцеральные признаки могут обнаруживаться в виде бронхо или ларингоспазмов, спастических запоров или гиперкинетических поносов, диспепсии, дискинезий желчного пузыря, болей в животе.
Хроническая магниевая недостача дополнительно проявляется пониженным тонусом мускулатуры, малой плотностью костной ткани.
Морфометрическая характеристика черепа при дисплазии соединительной ткани может изменяться из-за особенностей гемостаза. У больных зачастую диагностируются аортальные аневризмы, сопровождающиеся развитием хронического диссеминированного внутрисосудистого свертывания крови, как результата застоя в аневризменной полости и создания турбулентного тока в аорте. Возможно формирование ишемических поражений мозга, субарахноидальные, паренхиматозные кровоизлияния.
На сегодняшний день специалисты определили ряд фенотипических признаков дисплазии СТ. Их условно можно поделить на визуальные (те, которые можно заметить внешне) и на такие, которые обнаруживаются только по результатам тщательного внутреннего обследования.
У большинства пациентов наблюдается:
- высокая утомляемость, частая беспричинная усталость;
- частые простудные заболевания, ОРВИ;
- склонность к кровоточивости (большие потери крови при удалении зубов, при травмах, во время менструации у женщин);
- головокружения и боли в голове.
Более чем у 30% пациентов наблюдается так называемое «готическое небо», нарушения прикуса, гиперподвижность суставов, преждевременное старение лица, плоскостопие.
Боли при дисплазии соединительной ткани беспокоят в зависимости от того, какой орган поражен более других. Так, часто могут беспокоить периодические и недлительные боли в сердце, за грудиной и в области подреберья, спастические боли по ходу кишечника, головная боль. Неприятные болезненные ощущения в суставах появляются на этапе присоединения остеохондроза. Если имеются деформации грудной клетки или позвоночного столба, то боли в спине и груди возникают при продолжительном стоянии, ходьбе, либо даже в сидячем положении.
Страдают ли зубы при дисплазии соединительной ткани? Было проведено немало исследований, поскольку ученые пытались связать изменение качества зубной эмали с дисплазией соединительной ткани, что позволило бы более точно устанавливать диагноз заболевания. По итогам таких работ были обнаружены нарушения минерализации и формирования эмали зубов у пациентов с признаками соединительнотканной дисплазии. Это обусловлено недостаточной плотностью упаковки эмалевых призм на единицу объема. Кроме этого, призмы хаотично расположены, а органический матрикс слабо организован и минерализован. Склонность к неправильному развитию зубов и вероятность связанных с этим патологий определяется индивидуально, поскольку проявляется не у всех больных данным заболеванием.
Читайте также: