Геномные болезни. Особенности микроделеционных и дупликационных синдромов
Добавил пользователь Валентин П. Обновлено: 09.01.2026
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Синонимы русские
Микроделеции и микродупликации.
Синонимы английские
Microdeletion and microduplication.
Метод исследования
Полимеразная цепная реакция, фрагментный анализ, MLPA.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Микродупликационные/микроделеционные синдромы (ММС) представляют собой гетерогенную группу генетических нозологий, характеризующихся изменением количества копий участка хромосом. Синдромы данной группы чаще всего характеризуются нарушением когнитивных функций, задержкой речевого развития и задержкой роста, различного рода стигмами, дисморфизмами и мальформациями, затрагивающими широкий спектр систем и органов. Считается, что ММС являются одной из главных причин синдромального и несидромального спорадического отставания в развитии.
Исследование на основные формы микроделеционных и микродупликационных синдромов позволяет комплексно и одновременно выявлять 26 синдромов, такие как синдром ДиДжорджи, синдром Прадера - Вилли, синдром Ангельмана, синдром кошачьего крика, 1р36 делеция и другие. Рекомендуется проводить данное исследование в качестве первичного скрининга у пациентов с синдромальными и несиндромальными формами отставания в развитии.
Данный тест детектирует мутации, характерные для следующих нозологий: 1р36 микроделеционный синдром, 2p16.1-p15 микроделеционный синдром, 2q23.1 микроделеционный/микродупликационный синдром, 3q29 микроделеционный/микродупликационный синдром, 9q22.3 микроделеционный синдром, LIS1-ассоциированная лиссэнцефалия (синдром Миллера - Дикера / изолированная лиссэнцефалия / синдром двойной коры), SATB2 - ассоциированный синдром, нейрофиброматоз 1-го типа, KANSL1-связанная умственная отсталость, синдром Виттевеена - Колька, синдром Вольфа - Хиршхорна, синдром ДиДжорджи / велокардиофациальный синдром, синдром дупликации 15q, синдром дупликации гена MECP2, синдром кошачьего крика, синдром Лангера - Гидеона (трихоринофалангеальный синдром 2-го типа), синдром Прадера - Вилли / синдром Ангельмана, синдром Рубинштейна - Тейби, синдром Смита - Магениса, синдром Сотоса, синдром Фелана - МакДермида, синдром Вильямса - Бойрена, синдром Потоцки - Лупски, синдром Клайнфельтера, синдром Шерешевского - Тернера, синдром тройной Х хромосомы.
Для чего используется исследование?
- Диагностика микроделеционных и микродупликационных заболеваний
Когда назначается исследование?
- При подтверждении причин отставания и задержки развития.
- При дифференциальной диагностике причин пороков развития сердца, печени, почек, нервной системы.
- При подтверждении диагноза "микроделеционный" или "микродупликационный синдром".
Что означают результаты?
Наличие патогенной микроделеции или микродупликации подтверждает диагноз "ММС".
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды и индивидуальных генетических факторов.
Дупликации
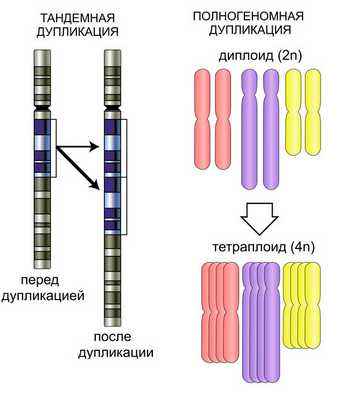
Представляют собой разновидность хромосомных перестроек, при которых происходит удвоение сегмента хромосомы. Если дупликация локализуется непосредственно за исходным участком хромосомы, то говорят о тандем-дупликации. Удвоенный сегмент может располагаться и в других участках. Удваиваться также могут крошечные сегменты (микродупликация).
Большинство хромосомных перестроек летальны. Они могут возникать вследствие неравного кроссинговера, ретротранспозиции, ошибок в гомологичной рекомбинации. Повторяющиеся участки в одной хромосоме могут размещаться как прямые или инвертированные тандемные повторы. Многократное повторение участка хромосомы называют амплификацией.
Генетические заболевания, обусловленные дупликациями
Структурные хромосомные мутации могут вызывать следующие синдромы:
- дупликации короткого плеча 9-й хромосомы. Признаки: выступающая верхняя губа и верхняя челюсть, гипертелоризм, широкий и круглый кончик носа, антимонголоидный разрез глаз, микробрахицефалия, анофтальм, аномальная форма ушных раковин. Сопровождается задержкой костного роста, олигофренией, аномалиями почек;
- трисомии длинного плеча 14-й хромосомы. Синдром обусловлен удвоением участка 14-й хромосомы и другими перестройками. Для больных характерно наличие микроцефалии, луковицеобразного носа, тонкой верхней губы, микростомии, опущенных углов рта, низко расположенных повернутых назад ушных раковин, короткой шеи, косолапости, пороков сердца;
- Видемана-Беквита. Развивается при дупликации короткого плеча 11-й хромосомы. У больных обнаруживают макроглоссию, макросомию, выступающий затылок, аномалии прикуса. Они страдают от дефектов межжелудочковых перегородок, добавочной селезенки, цитомегалии коры надпочечников, незавершенного поворота кишечника.
Микродупликация
Аномалия заключается в наличии дополнительной копии крошечной части хромосомы. Нарушение можно выявить с помощью таких методов, как FISH или хромосомный микроматричный анализ. Такая хромосомная перестройка наблюдается при болезни Шарко-Мари-Тута типа 1 А, при синдроме микродупликации 22q11.2 и т. д.
Пройти исследования на предмет структурных хромосомных аномалий можно в медико-генетическом центре «Геномед».
Микроделеционные и микродупликационные синдромы
Микроделеционные и микродупликационные синдромы являются заболеваниями, вызванными ультрамикроскопическими делециями или дупликациями функционально сцепленных генов на определенных участках хромосом. Постнатально диагноз предполагается на основании клинических проявлений и подтверждается предпочтительно хромосомным микроматричным анализом или флуоресцентной гибридизацией in situ.
Синдромы микроделеции лучше изучены, чем синдромы микродупликации, значение многих микродупликаций до сих пор не известно. В последние годы были более четко определены взаимные дупликации таких хорошо известных микроделеций, как 22q11.2 и 7q11.23.
Микроделеционные синдромы отличаются от синдромов хромосомной делеции Синдромы хромосомных делеций Данная группа заболеваний является результатом потери частей хромосом (делеций). Они могут стать причиной тяжелых врожденных аномалий, а также значительных умственных и физических отклонений. Прочитайте дополнительные сведения , при которых хромосомные делеции, как правило, заметны во время определения кариотипа из-за их большего размера (обычно > 5 мегабаз), в то время как аномалии при микроделеционных синдромах затрагивают более мелкие сегменты (обычно 1-3 мегабазы) и обнаруживаются только с помощью флуоресцентных зондов (флуоресцентной гибридизации in situ), а также хромосомного микроматричного анализа. Тот или иной генный сегмент может быть удален и дублирован (так называемая обратная дупликация). Клинический эффект микроскопической обратной дупликации как правило, одинаков, однако менее сильный, чем у делеций, которые включат в себя одинаковый сегмент. Термин синдром смежного гена, как правило, относится к состоянию, которое обычно связано с микроделециями, но также может быть связано с микродупликациями, при которых гены сгруппированы вместе. (См. также Технологии секвенирования следующего поколения Технологии генетической диагностики Технологии генетической диагностики быстро улучшаются. Небольшое количество ДНК может быть амплифицировано с помощью процесса полимеразной цепной реакции (ПЦР), которая позволяет производить. Прочитайте дополнительные сведения .)
Большинство клинически значимых микроделеций и микродупликаций, вероятно, возникают нерегулярно; однако, родителям с легким нарушением диагноз может быть поставлен, когда было проведено пренатальное тестирование после обнаружения того, что у ребенка есть аномалия.
Были выявлены многочисленные синдромы микроделеции с широкой вариацией проявлений болезни ( Примеры микроделеционных синдромов Примеры микроделеционных синдромов ).
Реципрокная микродупликация с участием хромосомы 17p11.2 ассоциирована с синдромом Потоцки-Лупски. Младенцы с этим расстройством имеют гипотонию, проблемы с питанием, отсутствие прибавки в весе, пороки сердца, задержку развития и аутизм.
Что такое микроделеционный синдром?
Несколько десятков лет назад, любой микроделеционный синдром воспринимался как патология неизвестного происхождения и не рассматривался как хромосомное отклонение. Дело в том, что у ученых не было возможности провести тонкую и точную диагностику наследственных заболеваний, не навредив здоровью матери и не повлияв на процесс развития плода.
Благо сегодня, для установления причины «сбоя» не требуется проводить забор плодных тканей или околоплодных вод. Ведь генетикам удалось установить, что с 8 недели беременности, в крови беременной женщины появляются фрагменты фетальной ДНК, способные дать огромный массив данных о возможных отклонениях в развитии будущего ребенка!
Синдром микроделеции — что это?
В отличие от моно- или трисомии, микроделеции приводят к уничтожению мелких фрагментов хромосом, провоцируя различные генетические аномалии. К списку самых распространенных заболеваний, подпадающих под группу микроделеционных синдромов, можно отнести:
- Делецию хромосомы 1p36, характеризующуюся пороками развития, умственными отсталостями, задержкой роста, внезапными судорогами, нарушениями работы органов чувств, дисморфизм черепно-лицевой области;
- Болезнь Вольфа-Хиршхорна, провоцирующую развитие сердечной и почечной недостаточности, задержкой развития, микроцефалией, нарушениями черт лица и т.д.;
- Синдром кошачьего крика, характеризующий отставанием в развитии, низкой массой при рождении, выраженной мышечной гипотонией. Основным признаком является характерный плач, напоминающий мяуканье кошки в результате изменения гортани;
- Болезнь Сотоса, выраженная высокорослостью, ускоренным ростом (до 4-5 лет), нарушением координации движения, тремором и судорожными приступами. Пациенты подвержены развитию онкологии.
В общей сложности, существует более 20 заболеваний, характеризующихся отсутствием отдельных фрагментов хромосом.
Основные причины возникновения
По результатам исследований, из 25 детей, родившихся с признаками, свойственными делеции, каждый 2-й ребенок унаследует болезнь от родителей. Из остальных, 40% зафиксированных случаев не имеют связи с наследственностью, в то время как у оставшихся 10% установить причины проявления болезней невозможно. Более того, некоторые заболевания (например - синдром Ди Джорджи) диагностируются в довольно позднем возрасте, из-за чего человек может жить с заболеванием, даже не догадываясь об этом.
Последствия хромосомного отклонения
К сожалению, большинство неинвазивных пренатальных тестов ДНК нацелены на выявление трисомии 13, 18, 21 хромосом и неспособны выявить микроделеционные аномалии. А ведь своевременная диагностика позволяет установить причину появления болезни и разработать эффективный способ её решения: от коррекции рациона питания, до работы над социализацией больного, страдающего от задержки развития.
В отсутствие своевременной диагностики и адекватного лечения, начальные симптомы могут развиться в более тяжелую форму, снижая не только качество, но и продолжительность жизни людей, страдающих от подобных недугов.
С недавних пор, жители нашей страны получили возможность пройти анализ ДНК на предмет отсутствия генетических мутаций, передающихся по наследству. Ведь в распоряжении специалистов лаборатории INLABgenetics имеется передовое материально-техническое оснащение, позволяющее выделять необходимые маркеры безболезненным и абсолютно безопасным способом: образцом для дородового генетического скрининга служит венозная кровь матери.
Для получения дополнительной информации об услуге достаточно позвонить по номеру «горячей линии» или оставить запрос на e-mail: менеджеры нашей компании готовы ответить на любой интересующий вас вопрос!
Читайте также: