Глаза при синдроме Сениора-Локена (Senior-Loken syndrome, SLS)
Добавил пользователь Morpheus Обновлено: 28.01.2026
Губчатая почка - довольно редкая врождённая аномалия, при которой кистозно расширены собирательные трубочки пирамид. Расширение, как правило, неравномерное; по ходу собирательных трубочек имеются дивертикулы, формирующие кистоподобные образования, и настоящие мелкие кисты, локализующиеся в мозговом слое почки. Почечные клубочки, другие элементы нефрона и лоханочно-чашечная системы развиты нормально.
Этот вид порока развития структуры почки впервые описал Дуззи в 1939 г. Он же предложил термин "губчатая почка".
Эпидемиология
Губчатую почку встречают достаточно редко, почти исключительно у лиц мужского пола, чаще всего порок бывает двусторонним.
Порок развития наследуется по частично сцепленному с полом типу.
Аномалия обычно ничем клинически не проявляется, если не развиваются осложнения. Однако у половины носителей этой аномалии и в неосложнённых случаях отмечают постоянную умеренную протеинурию, микрогематурию или лейкоцитурию.
Пиелонефрит губчатой почки отличается упорным течением, склонностью к рецидивам и может привести к почечной недостаточности. Клинические проявления становятся поводом для проведения диагностического обследования.
Диагноз ставят на основании данных экскреторной урографии по типичному признаку ("букет цветов" в зоне пирамид).
При отсутствии осложнений лечения не требуется. При осложнениях может понадобиться лечение пиелонефрита, длительное и упорное.
К оперативному лечению прибегают в случае наличия камней, обтурирующих мочевыводящие пути, при кровотечении или гнойном пиелонефрите губчатой почки.
Cиндром Сениора-Локена
Синонимы: нефронофтиз с дистрофией сетчатки, синдром почечная дисплазия-аплазия сетчатки, синдром Сеньора-Локена
Определение и общие сведения
Cиндром Сениора-Локена является очень редким аутосомно-рецессивным заболеванием, характеризующимся ассоциацией нефронофтиза с дистрофией сетчатки.
Распространенность во всем мире оценивается примерно в 1/1 000 000 человек. Наследование аутосомно-рецессивное.
Этиология и патогенез
Cиндром Сениора-Локена представляет собой генетически гетерогенную цилиопатию. Описаны мутации в 7 разных генах: NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, SDCCAG8. Данные гены кодируют белки первичных ресничек, играющих ключевые роли в развитии и функционировании нескольких типов клеток, включая фоторецепторы сетчатки и эпителиальные клетки трубочек почек. Предполагается, что эпистатические взаимодействия, олигогенное наследование и модифицирующие аллели влияют на экспрессивность различных экстраренальных фенотипов патологии.
Клинические проявления
Cиндром Сениора-Локена проявляется сразу после рождения или в детстве симптомами нефронофтиза, такими как полиурия, полидипсия, вторичный энурез и анемия. Прогрессирование болезни может привести к развитию острой или хронической почечной недостаточности и в итоге к терминальной стадии почечной недостаточности. Патология зрения включают в себя врожденную или раннюю выраженную потерю зрения из-за дистрофии сетчатки. В редких случаях могут наблюдаться также другие дополнительные клинические симптомы, такие как фиброз печени, ожирение и неврологические расстройства.
Диагностика
Рекомендуются полное офтальмологическое обследование - флюороскопия, тест на остроту зрения, дефекты преломления, тесты на цветное зрение, окулярную подвижность и электроретинограмма), а также обследование почек - функция почек, анализ мочи и УЗИ брюшной полости. Также рекомендуется исключить фиброз печени (анализ функции печени и УЗИ) и проведение неврологического обследования для младенцев. Генетическая диагностика требует скрининга мутаций вовлеченных генов, делеция гена NPHP1 является самой частой аномалией.
Дифференциальный диагноз
Cиндром Сениора-Локена имеет генетическое и клиническое совпадение с другими цилиопатиями, в частности с изолированными нефронофтизом, заболеваниями, связанными с синдромом Жубера, главным образом с синдромом Жубера с окулоренальным дефектом, синдромом Барде-Бидля и синдромом Альстрема.
Пациенты должны регулярно контролироваться педиатром-нефрологом - мониторинг веса и роста, контроль артериального давления, исследования функции почек, измерение концентрации мочи и анализ выведения натрия почками. Раннее лечение нефронофтиза может задержать прогрессирование почечной недостаточности и минимизировать вторичные осложнения. В случае развития терминальной стадии почечной недостаточности пациенты нуждаются в диализе или трансплантации почки. В настоящее время нет способа предотвращения прогрессирующей потери зрения.
Прогноз зависит в основном от развития почечных осложнений, которые являются основной причиной смерти больных.
Нефронофтиз
Нефронофтиз - хроническая тубулоинтерстициальная нефропатия, которая прогрессирует до терминальной стадии почечной недостаточности.
Распространенность составляет 1/100 000 человек. Нефронофтиз передается аутосомно-рецессивно.
Было идентифицировано пять генов, ассоциированных с развитием нефронофтиза. Ген NPHP1 (2q13), гомозиготные делеции гена NPHP1 наблюдались у 70% пораженных детей, и их обнаружение с помощью ПЦР позволяет установить диагноз. Мутации гена NPHP2, кодирующие инверсин, ответственны за инфантильную форму нефронофтиза, которая прогрессирует до конечной стадии почечной недостаточности до достижения пациентом 5-летнего возраста. Мутации гена NPHP3 (3q21-22) описаны в большой венесуэльской семье и вызывают позднюю начальную форму заболевания. Мутации гена NPHP4, локализованные в хромосоме 1p36, наблюдались в нескольких семействах, у некоторых из которых отмечались вовлечение сетчатки. Мутации гена IQCB1/NPHP5, локализованного на хромосоме 3q13, недавно были идентифицированы у пациентов с аутосомно-рецессивным нефронофтизом и синдромом Сениора-Локена.
Болезнь является клинически и генетически гетерогенной. Описаны три основные клинические формы нефронофтиза.
Юношеский нефронофтиз, наиболее частая форма, прогрессирует до конечной стадии почечной недостаточности в возрасте до 15 лет и отвечает за 15% случаев почечной недостаточности у детей. Первые признаки патологии появляются после 2-го возраста с дефектом концентрации мочи, приводящим к полиурию и полидипсии, отставанию в физическом развитиии, прогрессирующим ухудшением функции почек без признаков гломерулярной болезни. Ультрасонография почек выявляет почки нормального размера. Гистологические поражения затрагивают базальные мембраны трубочек, которые нерегулярно утолщены, многослойны или истончены. Также присутствует интерстициальный фиброз. У некоторых детей наблюдаются экстраренальные симптомы: тапеторетинальная дегенерация (как в случае синдрома Сениора-Локена), интеллектуальный дефицит, мозжечковая атаксия, аномалии костей или вовлечение печени.
Инфантильный нефронофтиз представляет собой хроническую тубулоинтерстициальную нефропатию с кортикальными микроцистами, прогрессирующую до терминальной стадии почечной недостаточности в возрасте до 5 лет.
Поздняя форма нефронофтиза является более редкой формой заболевания. Клинические и гистологические признаки сходны с таковыми у подростков, но возраст, при котором почечная недостаточность достигает терминальной стадии, составляет, в среднем, 19 лет.
Антенатальная диагностика нефронофтиза может быть выполнена, если мутация была идентифицирована у одного ребенка в семье.
В настоящее время нет адекватной терапии нефронофтиза для предотвращения прогрессирования почечной недостаточности.
Сениора-Локена синдром
Сениора-Локена синдром 1 - является аутосомно-рецессивным заболеванием, основными клиническими признаками которого является сочетание патологии почек (нефронофтиз) и глаз (пигментная дегенерация сетчатки). Распространенность нефронофтиза оценивается в среднем 1 на 100000 человек, причем у 1 из 10 больных, также наблюдается патология сетчатки, то есть синдром Сениора-Локена. Выделяют несколько форм синдрома, кодирующихся разными генами (см. Таблицу 1). Синдром Сениора-Локена 1 обусловлен делецией гена NPHP1, расположенного на хромосоме 2, в гомозиготном состоянии.
Нефронофтиз характеризуется нарушением тубулярных функций почек, что проявляется снижением концентрационной функции почек, развитием полиурии, полидипсии, синдрома потери солей (гипонатриемия, гипокальциемия и гипокалиемия). При прогрессировании заболевания развивается хроническая почечная недостаточность (ХПН). Терминальная ХПН наступает обычно после на втором десятке лет жизни. Для синдрома Сениора-Локена характерно раннее прогрессирующее поражение сетчатки. Однако, дети могут быть слепыми от рождения.
| Форма заболевания | OMIM | Локус | Ген |
| SLSN1 | 266900 | 2q13 | NPHP1 |
| SLSN3 | 606995 | 3q22 | не известен |
| SLSN3 | 606996 | 1p36 | NPHP4 |
| SLSN5 | 609254 | 3q21 | NPHP5/IQCB1 |
| SLSN6 | 610189 | 12q21 | NPHP6/CEP290 |
| SLSN7 | 613615 | 1q44 | SDCCAG8 |
| SLSN8 | 616307 | 4p14 | WDR19 |
| SLSN9 | 616629 | 2q37 | TRAF3IP1 |
Ген NPHP1 кодирует белок нефроцистин-1. Этот белок, играет важную роль в формировании так называемых цилий. Цилии (реснички или жгутики) - это органеллы, представляющие собой микроскопические пальцевидные выступы, расположенные на апикальной поверхности эукариотических клеток. Они обеспечивают течение различных тканевых жидкостей, выполняют сенсорные функции или участвуют в сигнальных путях, передающих информацию внутри и между клетками. Это необходимо для нормального функционирования многих типов клеток и тканей. Белок нефроцистин-1 находится у основания ресничек в клетках почек, дыхательных путей, светочувствительных клеток сетчатки глаза и др. И хотя точные функции нефроцистина-1 еще не изучены, предполагается, что он взаимодействует с рядом других белков в составе большого белкового комплекса, обеспечивающего работу ресничек.
В 2009 году Khanna H. с соавторами показали, что модификатором дегенерации сетчатки у пациентов с цилиопатиями в том числе и с синдромом Сениора-Локена, может быть распространенный аллель гена RPGRIP1L с.685А (с. 685G>A, р.A229T). Этот аллель ассоциирован с потерей фоторецепторов, что ведет к дегенерации сетчатки.
В Центре молекулярной генетики проводится прямая ДНК-диагностика синдрома Сениора-Локена которая основана на поиске делеции в гене NPHP1 (анализ числа копий гена NPHP1) методом количественной мультиплексной лигазной реакции с последующей амплификацией (MLPA-PCR-анализ).
Синдром CLC ( Синдром Клерка-Леви-Кристеско , Синдром Лауна-Ганонга-Левайна )
Синдром CLC - это один из вариантов преждевременного возбуждения желудочков, который обусловлен наличием добавочного пучка Джеймса. Болезнь характеризуется ускоренным проведением нервных импульсов от предсердий к АВ-узлу, укорочением интервала PQ и неизмененным комплексом QRS. У большинства людей синдром протекает в скрытой форме, клинические признаки возникают только при осложнении заболевания пароксизмальной тахикардией. Для диагностики болезни выполняют ЭКГ, ЭхоКГ, чреспищеводное ЭФИ. Профилактическое лечение аномалии CLC проводят антиаритмическими препаратами, при рефрактерных формах заболевания назначается радиочастотная абляция.
МКБ-10



Общие сведения
Синдром CLC (Клерка-Леви-Кристеско) получил свое название благодаря французским кардиологам, которые впервые описали его симптоматику в 1938 году. Аббревиатура CLC используется в континентальной Европе и России. В США и Великобритании болезнь чаще называют синдромом Лауна-Ганонга-Левайна (LGL) в честь американских кардиологов, которые изучали механизмы преждевременного возбуждения миокарда. Патология обнаруживается у 0,5% взрослого населения, однако она не всегда себя проявляет и нередко диагностируется случайно при обследовании по другим показаниям.

Причины
Синдром CLC - врожденная аномалия развития сердца, молекулярно-генетические основы которой недостаточно изучены в современной кардиологии. При этом заболевании в проводящей системе сердца формируется дополнительный пучок Джеймса. Его волокна соединяют синусовый узел и нижнюю часть атриовентрикулярного узла, продолжаются дальше до пучка Гиса.
Патология развивается в первом триместре беременности, на 5-6 неделе, когда образуются проводящие пути в сердце. Хотя формирование сердечной аномалии связывают с разнообразными тератогенными влияниями, роль факторов внешней среды в развитии заболевания на сегодня не доказана. Исследовательские данные позволяют предположить наследственную предрасположенность синдрома CLC у некоторых пациентов.
Патогенез
При синдроме CLC нервные импульсы проходят через атриовентрикулярный узел, не задерживаясь в нем, и вызывают возбуждение миокарда желудочков. Сократимость сердечной мышцы регулируется системой Гиса-Пуркинье, поэтому все отделы сердца деполяризуются одновременно, как при физиологическом ритме. Заболевание проявляется сокращением интервала PQ - основной признак преждевременного возбуждения, сочетающийся с нормальным размером желудочковых комплексов.
Часть исследователей подчеркивает роль добавочных трактов Брехенмахера в патогенезе заболевания. Эти волокна определяются намного реже - всего у 0,03% людей с аномалией CLC. Они дают дополнительный обходной путь для нервных импульсов: проводят их от предсердия к пучку Гиса и заметно ускоряют процессы возбуждения миокарда. Высказывают мнение, что наличие этих добавочных пучков отягощает прогноз для пациента.
Главная особенность синдрома CLC, которая важна для практической кардиологии, - участие добавочных проводящих путей Джеймса и Брехенмахера в патологических петлях re-entry. Наблюдается круговая циркуляция возбуждающих импульсов, которая нарушает нормальный сердечный ритм и провоцирует пароксизмальные тахикардии. Такое состояние называют «феноменом предвозбуждения».
Симптомы
Клинические проявления синдрома CLC развиваются при осложнении болезни тахиаритмией. В этом случае больные предъявляют жалобы на учащенное сердцебиение, одышку, дискомфорт в левой половине грудной клетки. Возможно развитие головокружения, тошноты, предобморочного состояния. Изредка пациенты жалуются на сжимающие боли за грудиной, вздутие живота, рвоту. Длительные пароксизмы тахикардии заканчиваются обильным мочеиспусканием.
Приступы тахикардии в основном кратковременны, исчезают самостоятельно, не вызывают критических нарушений. При резком ухудшении здоровья пароксизмы удается прекратить немедикаментозными методами: натуживанием на вдохе, опусканием лица в холодную воду, задержкой дыхания. Такие методы называют вагусными пробами, они основаны на активации парасимпатических нервных влияний, регулирующих сердечную деятельность.
Осложнения
Основная проблема аномалии CLC - риск развития жизнеугрожающих тахиаритмий, которые без экстренного лечения могут заканчиваться летально. Вероятность нарушений ритма повышается у пациентов с сопутствующими пороками сердца, хроническими кардиальными патологиями (ИБС, артериальная гипертензия). В группе риска по развитию осложнений находятся профессиональные спортсмены, военные, сотрудники экстренных служб, которые работают в условиях физического и морального напряжения.
Диагностика
Большинство пациентов с синдромом CLC не знают о существовании у себя такой болезни и не обращаются к врачам. Сердечную аномалию обнаруживают при плановом проведении ЭКГ на профосмотре, при визите к врачу-кардиологу с другими жалобами. Клиническая картина болезни скудна, поэтому для определения преждевременного сокращения миокарда желудочков и постановки нозологического диагноза назначают следующие исследования:
- ЭКГ. При синдроме CLC на электрокардиограмме определяется укорочение интервала PQ менее 0,11 с, нормальная ширина комплекса QRS (до 0,12 с) без добавочной дельта-волны в начале зубца. При сопутствующей блокаде ножек пучка Гиса обнаруживается расширение и деформация QRS, характерные М-образные зубцы. Для уточнения диагноза назначается холтеровское мониторирование.
- УЗИ сердца. Эхокардиография применяется для оценки структурно-функциональных особенностей миокарда, исследования сердечных камер, исключения сопутствующих врожденных пороков. Сократительную способность миокарда оценивают по фракции выброса.
- Чреспищеводное ЭФИ. Электрофизиологическое исследование сердца выполняется, чтобы обнаружить добавочные пути проведения импульсов и подтвердить синдром преждевременного возбуждения. Методика показывает количество и локализацию дополнительных пучков, их функциональность.
- Рентгенография ОГК. Рентгенологическая диагностика показывает контуры сердца, позволяет предположить структурные аномалии и перестройки на фоне хронической патологии. Также по данным рентгеновских снимков исключают сопутствующую легочную патологию.
- Лабораторные методы. Кардиологическим пациентам назначают стандартный комплекс анализов, который включает клиническое и биохимическое исследование крови, определение показателей липидограммы, изучение свертывающей системы крови. По показаниям проводят анализ острофазовых белков.
Дифференциальная диагностика
При постановке диагноза синдром CLC дифференцируют с другой типичной причиной преждевременного желудочкового возбуждения - синдромом Вольфа-Паркинсона-Уайта (WPW). Дифференциальная диагностика требует детального изучения клинической картины и тщательной оценки ЭКГ для обнаружения дельта-зубцов. В периоде приступа CLC необходимо исключить блокаду пучка Гиса, острый коронарный синдром.
Лечение синдрома CLC
Консервативная терапия
Пациентам редко требуется экстренная терапия, поскольку заболевание не сопряжено с высоким риском кардиоваскулярных кризов. Для контроля работы сердца назначаются противоаритмические препараты длительными курсами. В основном используются бета-адреноблокаторы, сердечные гликозиды, блокаторы кальциевых каналов. Цель назначения медикаментов - профилактика пароксизмов наджелудочковой тахиаритмии.
Хирургическое лечение
При неэффективности медикаментозной терапии и частых повторениях приступов тахикардии предпочитают проводить радиочастотную абляцию. Это малоинвазивный метод лечения, с помощью которого разрушают патологические проводящие пути и устраняют их негативное влияние на сердечный ритм. Результативность однократной процедуры составляет 92-95%, по показаниям ее проводят повторно для достижения 100% эффекта.
Прогноз и профилактика
У пациентов с аномалией CLC при отсутствии клинических признаков заболевания прогноз благоприятный. Они имеют минимальный риск развития пароксизмальной тахикардии, не нуждаются в постоянном диспансерном наблюдении. Если на фоне синдрома Клерка-Леви-Кристеско возникают нарушения сердечного ритма, отдаленный прогноз зависит от своевременности диагностики и успешности лечения. Поскольку причины формирования аномальных проводящих путей не установлены, меры профилактики отсутствуют.
3. Феномены преждевременного возбуждения желудочков/ М.В. Потапова, О.Р. Соколова// Вестник современной клинической медицины. - 2010. - №2.
Синдром Сусака ( Ретино-кохлео-церебральная васкулопатия )
Синдром Сусака — это редкое аутоиммунное заболевание с триадой клинических синдромов: энцефалопатией, тугоухостью, микроангиопатией сетчатки. Симптоматика включает нарушения внимания, памяти, поведения, ухудшение слуха и вариабельные зрительные расстройства. Диагноз устанавливается по результатам церебральной МРТ, флюоресцентной ангиографии сетчатки, аудиометрии. Наиболее эффективным считается интенсивное иммуносупрессорное лечение комбинацией глюкокортикоидных и цитостатических препаратов с добавлением внутривенного интерферона и терапии моноклональными антителами к В-лимфоцитам.




Синдром Сусака является аутоиммунной патологией, точные причины развития которой пока не определены. Генетических или наследственных связей в возникновении заболевания не выявлено. Как и для большинства аутоиммунных процессов, среди этиологических факторов, приводящих к дисфункции иммунной системы, выделяют перенесенные вирусные инфекции, перестройку организма матери в период беременности, длительную терапию гормональными фармпрепаратами. Указанные триггеры способны провоцировать выработку аутоантител, что вызывает развитие васкулопатии Сусака.
Основным патогенетическим механизмом заболевания в настоящее время считается дисфункции отдельных звеньев иммунной системы, приводящая к выработке антител против собственных эндотелиальных клеток, выстилающих стенки сосудов мелкого калибра (артериол, капилляров) во внутреннем ухе, головном мозге и сетчатке. Повреждение эндотелия сосудов микроциркуляторного русла с развитием микроангиопатии влечет за собой их сужение и окклюзию с нарушением микроциркуляции. Результатом является недостаточное кровоснабжение (ишемия) и кислородное голодание (гипоксия) окружающих тканей.
Изменения в головном мозге характеризуется очагами микроинфарктов, повреждающих нейроны, их отростки и миелиновую оболочку. В таких местах нервная ткань замещается глиальной, происходит формирование мелких очагов глиоза, что приводит к энцефалопатии. Эндотелиопатия ретинальных сосудов сопровождается снижением зрения.
Расстройство микроциркуляции во внутреннем ухе приводит к ухудшению слуха. До конца не исследован вопрос избирательной локализации сосудистых изменений. Предрасположенность к триаде симптомов при синдроме Сусака возможно обусловлена тем, что микрососудистые окклюзии в головном мозге, сетчатке и улитке вызывают более выраженные симптомы, чем в других местах.
Характерная триада клинических проявлений синдрома Сусака включает нарушения со стороны головного мозга (энцефалопатию), снижение слуха, зрения. Однако у 80% заболевших патология манифестирует моноорганным поражением. Начало заболевания проявляется интенсивными головными болями мигренозного характера, локализующимися то в одной, то в другой половине головы. На высоте боли возможна тошнота, светобоязнь. Головная боль считается наиболее частым продромальным симптомом и может появиться за несколько месяцев до развития энцефалопатии.
В некоторых случаях синдром дебютирует явными признаками энцефалопатии: психическими расстройствами, когнитивными изменениями, ухудшением памяти. Нарушения психики варьируют от депрессивных состояний до агрессивного поведения. Возможны параноидальные расстройства, характеризующиеся наличием навязчивой бредовой идеи и связанных с ней изменений в поведении: повышенной подозрительности, раздражительности, обидчивости или враждебности. Поскольку восприятие действительности у заболевшего искажено, указанные изменения могут быть замечены только его близкими.
Расстройство когнитивной сферы проявляется рассеянностью, затруднениями при концентрации внимания, сложностями с запоминанием текущей информации, потерей памяти (амнезией). Прогрессирование васкулопатии Сусака сопровождается появлением очаговой неврологической симптоматики: мышечной слабостью в верхней конечности (монопарез) и дизартрией. Больной не может полноценно двигать рукой, удерживать в ней предметы. Речь его становится невнятной и малопонятной окружающим. Отмечается спутанность сознания, развивается деменция (слабоумие). В ряде случаев наблюдаются судороги.
Повреждение сосудов внутреннего уха проявляется понижением слуха (тугоухостью), носящим односторонний или асимметричный двусторонний характер. Затруднено восприятие преимущественно низко- и среднечастотных звуковых колебаний, распространена плохая разборчивость речи. Тугоухость сопровождается звоном в ухе, сочетается с вестибулярными нарушениями: головокружением, дискоординацией, нистагмом (непроизвольными подергиваниями глазных яблок). Присоединение симптомов слуховой и вестибулярной дисфункции усугубляет психические расстройства.
Выраженность зрительной дисфункции зависит от локализации и распространенности ретинальной васкулопатии. Пациенты отмечают фотопсии — периодические вспышки или «блестящие шары» в поле зрения. Типичны выпадения отдельных участков поля зрения (скотомы), описываемые больными как «пятна», в которых окружающее видно расплывчато. Синдром Сусака с обширным поражением артериол сопровождается атрофией зрительного нерва и значительным сужением полей зрения. При локализации микроциркуляторных нарушений исключительно в периферических отделах сетчатки, их клинические проявления могут отсутствовать.
Прогрессирующее течение васкулопатии ведет к выраженным патологическим изменениям в тканях головного мозга с развитием грубых психических симптомов и очаговых нарушений в виде парезов, затрудняющих самообслуживание больного. Усугубление микроциркуляторных расстройств сетчатки может привести к полной слепоте. Осложнением тугоухости выступает полная утрата слуха. Быстрая и ранняя терапия во многих случаях позволяет остановить прогрессирование, предупредить развитие тяжелых нарушений психики, необратимых изменений сетчатки и органа слуха.
Дебют синдрома Сусака одним из характерной триады признаков значительно затрудняет его раннюю диагностику. Выявление очаговой симптоматики (нистагма, пареза, дизартрии) и когнитивных изменений помогает врачу-невропатологу заподозрить органический характер имеющихся психических нарушений и диагностировать энцефалопатию. К обследованию пациента наряду с неврологом привлекаются лор-специалисты и офтальмологи. Ключевое значение в диагностике имеют следующие исследования:
- Анализ цереброспинальной жидкости. Материал для анализа получают путем спинномозговой пункции. В исследуемом материале выявляется умеренно повышенное содержание белка и лимфоцитарный плеоцитоз — увеличенное содержание клеток преимущественно за счет лимфоцитов.
- МРТ головного мозга. Множественные мелкоочаговые поражения наблюдаются в мозолистом теле, внутренней капсуле, перивентрикулярном белом веществе, стволе, мозговых и мозжечковых ножках, базальных ганглиях и таламусе. Патогномоничным для синдрома Сусака признаком является типичная форма очагов поражения в виде «жемчужного ожерелья» или «снежинок», выявляемая на Т2-взвешенном изображении. При исследовании с контрастным усилением отмечается более интенсивная окраска свежих очагов.
- Периметрия. Возможно выявление сужения полей зрения, выпадения различных квадрантов, наличия центральных и парацентральных скотом.
- Офтальмоскопия. Исследование глазного дна может показать окклюзию ветвей центральной ретинальной артерии. Когда окклюзии ограничены небольшими периферическими артериолами, глазное дно выглядит нормальным.
- Флюоресцентная ангиография. Является лучшим методом для выявления окклюзий артериол сетчатки. Множественные окклюзии и очаги «затекания» флуоресцеина — патогномоничные критерии васкулопатии Сусака, диагностируемые даже у пациентов, не имеющих клинически выраженных офтальмологических симптомов. Макулярная область в большинстве случаев остается интактной.
- Оптическая когерентная томография. Используется в качестве диагностического инструмента для анализа морфологической целостности структур сетчатки. Позволяет дифференцировать синдром Сусака с типичным пятнистым истончением слоя нервных волокон от рассеянного склероза, при котором истончение носит диффузный характер.
- Тональная аудиометрия. Фиксирует сенсоневральную тугоухость в диапазоне низких и средних частот. Дополнительно рекомендовано исследование акустических вызванных потенциалов.
Синдром дифференцируют от демиелинизирующих заболеваний (рассеянного склероза, рассеянного энцефаломиелита) на основании данных церебральной МРТ и ангиографии сетчатки. Энцефалопатия требует дифференцировки от инфекционного энцефалита. При сочетании тугоухости с вестибулярной дисфункцией необходимо исключить болезнь Меньера.
Полиорганные расстройства также характерны для системных васкулитов, как синдром Когана, гранулематоз Вегенера. В отличие от васкулопатии Сусака, синдрома Когана сопровождается интерстициальным кератитом. При гранулематозе Вегенера тугоухость сочетается с поражением дыхательных путей и почек, что нетипично для синдрома Сусака.
МРТ головного мозга (А, В), фотография глазного дна (С) и ангиография глазного дна (D) при синдроме Сусака
Лечение синдрома Сусака
Пациентам рекомендована низкосолевая диета с высоким содержанием витаминов группы В, витамина С и К. В основе этиопатогенетической фармакотерапии лежит интенсивная иммуносупрессия, направленная на снижение продукции аутоантител, повреждающих сосудистый эндотелий. В практической неврологии комплексная терапия синдрома осуществляется путем индивидуального подбора нескольких медикаментов. Лечащий врач делает назначение в соответствии с клинической картиной, давностью и тяжестью заболевания, корректирует его на основании динамики симптомов. Основными группами применяемых препаратов являются:
- Глюкокортикоиды. Обладают иммуносупрессивными свойствами, оказывают противовоспалительный эффект. Важное значение имеет способность глюкокортикостероидов поддерживать метаболические процессы в организме на фоне хронического заболевания.
- Цитостатики. Характеризуются выраженным ингибирующим действием на иммунную систему. Несмотря на выраженные побочные эффекты, являются необходимым компонентом терапии васкулопатии Сусака любой степени тяжести.
- Внутривенные иммуноглобулины (ВВИГ). Выступают неспецифическими корректорами иммунитета. Иммуномодуляторный эффект связан с понижением продукции собственных (аутоиммунных) иммуноглобулинов и иммунных комплексов. ВВИГ позволяет путем заместительной терапии обеспечить противомикробный иммунитет у имеющих иммунодефицит пациентов.
- Генно-инженерные биологические препараты (ГИБП). Используются моноклональные антитела к антигенам зрелых В-лимфоцитов, ответственных за продукцию аутоантител. После связывания с В-лимфоцитом ГИБП инициирует его лизис, что влечет снижение количества образующихся антиэндотелиальных аутоантител. Гемопоэтические клетки и здоровые плазматические клетки остаются нетронутыми.
- Антитромботические средства. Назначаются пациентам с повышенным риском тромбообразования.
При тяжелом течении для очистки крови от циркулирующих в ней иммунных комплексов применяется плазмаферез. Возможно проведение сеансов гипербарической оксигенации для компенсации гипоксических процессов, обусловленных поражением сосудов.
Синдром Сусака имеет моноциклическое, полициклическое или хроническое непрерывное течение. При раннем начале терапии возможен хороший прогноз. Своевременная диагностика и раннее назначение иммуносупрессивной терапии приводят к почти полному выздоровлению, несмотря на значительную энцефалопатию при обращении пациента. Однако постановка диагноза обычно запаздывает, что влечет развитие осложнений. Приблизительно у 50% пациентов сохраняются когнитивные нарушения. Поскольку этиологические факторы аутоиммунной патологии не определены, реальные способы профилактики пока не разработаны.
1. Синдром Сусака (ретино-кохлео-церебральная васкулопатия)/ Бекетова Т.В., Коновалов Р.Н.// Научно-практическая ревматология. - 2018. - 56(2).
2. Синдром Сусака, обзор и описание клинического случая/ Вергунова И.Ю., Малкова Н.А., Коробко Д.С.// Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. - 2019. - 119(2-2).
3. Рассеянный склероз и синдром Сусака (дифференциальная диагностика, лечение, описание случая)/ Чухловина М.Л.// Труды Мариинской больницы. - 2010.
4. Diagnosis and classification of Susac syndrome/ García-Carrasco M, Mendoza-Pinto C, Cervera R.// Autoimmun Rev. - 2014. - Apr-May;13(4-5).
Цилиопатии
Цилиопатии — генетически обусловленные заболевания, возникающие при нарушении структуры или функции цилий.
Содержание
Структура и функции цилий
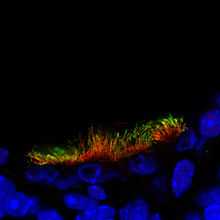

Пример флагелл, расположенных на поверхности эпителиальных клеток тимуса. Картинка получена при помощи иммунофлуоресцентного анализа.


Цилии (реснички или жгутики) представляют собой эволюционно консервативные органеллы, расположенные на апикальной поверхности эукариотических клеток. В зависимости от внутренней структуры и выполняемой функции все цилии можно разделить на два основных типа: подвижные (флагеллы) и неподвижные (сенсорные).
Исторически, флагеллы являются наиболее изученным типом цилий. Обычно расположенные в большом количестве на поверхности клеток, флагеллы создают волнообразное биение, которое, в свою очередь, обуславливает движение одиночных клеток и течение различных тканевых жидкостей. Ярким примером может служить течение цереброспинальной жидкости в желудочках головного мозга, а также движение яйцеклеток и сперматозоидов относительно соответствующего репродуктивного тракта. Внутренняя структура флагелл (аксонема) может быть охарактеризованна формулой (9+2), где девять периферически расположеных дублетов микротрубочек окружают две синглетные микротрубочки в центре. Наличие аксонемного динеина в структуре флагелл обеспечивает взаимное скольжение микротрубочек и, как следствие, волнообразное биение всей органеллы.
В отличие от флагелл, сенсорные цилии являются солитарными органеллами. Этот тип цилий не имеет центральных синглетных микротрубочек (9+0) и аксонемных динеинов. Вследствие этого, сенсорные цилии теряют способность к биению и в основном содержат различные рецепторные белки на своей поверхности, выполняя сенсорные фукции. Одним из примеров клеток с сенсорными цилиями являются палочки и колбочки сетчатки глаза.
Молекулярные механизмы цилиопатий
Цилиогенез (процесс образования цилий) может быть разделен на четыре основные стадии: формирование центриолей, миграция центриолей к апикальной мембране, трансформация центриолей в базальные тельца, формирование аксонемы и ассоциированных структур. Мутации генов, контролирующих данные процессы, в подавляющем большинстве случаев приводят к структурным аномалиям цилиий, и, как следствие, к развитию цилиопатий. Наиболее изученными цилиопатийными генами являются гены контролирующие процесс интрафлагеллярного транспорта. Отдельно стоит отметить гены, кодирующие функциональные протеины цилий. Мутации некоторых из них (например, полицистинов) также приводят к возникновению цилиопатий.
Цилиопатийные синдромы
В организме человека цилии присутствуют практически на любых типах клеток и выполняют как мотильные, так и сенсорные функции. Вследствие распространённости цилий, их структурные или функциональные дефекты вызывают широкий спектр патологий и синдромов (цилиопатий), включая:
- ( Alström syndrome )
- Синдром Барде — Бидля ( Bardet-Biedl syndrome )
- Синдром Сениора-Локена ( Senior-Løken syndrome )
- Синдром Жубера ( Joubert syndrome ) ( Meckel-Gruber syndrome ) ( Kartagener syndrome )
- Врожденный амавроз Лебера ( Leber’s congenital amaurosis )
- ОФД-синдром I типа ( Oral-facial-digital syndrome type I — OFD I )
- Синдром Жене ( Jeune syndrome )
- Синдром Эллис-ван-Кревельда ( Ellis van Creveld syndrome )
- Синдром Сенсебреннера ( Sensenbrenner syndrome )
- Синдром коротких рёбер — полидактилии ( Short rib — polydactyly syndrome )
- Синдром Фон Хиппеля-Линдау ( Von Hippel-Lindau syndrome ) ( Lowe syndrome )
Цилиопатии являются плейотропными патологиями и характеризуются такими клиническими симптомами, как поликистоз почек, печени и поджелудочной железы, деградация сетчатки глаза, асимметрия висцеральных органов, полидактилия, челюстно-лицевые дефекты и дефекты одонтогенеза, гидроцефалия, ожирение, бесплодие.
Читайте также: