Глухота с изменениями сетчатки, мышечной слабостью и умственной отсталостью
Добавил пользователь Дмитрий К. Обновлено: 21.01.2026
Категории МКБ: Другие формы умственной отсталости (F78), Умственная отсталость глубокая (F73), Умственная отсталость легкой степени (F70), Умственная отсталость неуточненная (F79), Умственная отсталость тяжелая (F72), Умственная отсталость умеренная (F71)
Общая информация
Краткое описание
Одобрен
Объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения Республики Казахстан
от «5» октября 2017 года
Протокол № 29
Умственная отсталость - группа различных наследственных, врожденных или рано приобретенных состояний общего психического недоразвития, состояние задержанного или неполного развития психики, которое в первую очередь характеризуется нарушением способностей, проявляющихся в период созревания и обеспечивающих общий уровень интеллектуальности, то есть когнитивных, речевых, моторных и социальных способностей. Синонимы У.о. - «общее психическое недоразвитие», «психическое недоразвитие», «олигофрения». В DSM-5 - термин У.о. заменен на «Интеллектуальная несостоятельность».
NB! Интеллектуальная несостоятельность включает в себя нарушения умственных способностей, которые оказывают влияние на адаптивное функционирование, в трёх зонах, или областях [3]:
· схематизация, включает в себя навыки овладения речью, чтением, письмом, математикой, аргументации, эрудиции и памяти;
· социализация, включает в себя навыки эмпатии, суждения об отношениях, общения, способности дружить и поддерживать дружбу, и подобные им способности;
· реальность, касается самоорганизации в вопросах заботы о себе, ответственности по работе, умения распоряжаться деньгами, восстанавливать свои силы, устраивать учебные и трудовые дела.
Код (ы) МКБ-10:
| МКБ -10 | |
| Код | Название |
| F-70 | Легкая умственная отсталость |
| F-71 | Умеренная умственная отсталость |
| F-72 | Тяжелая умственная отсталость |
| F-73 | Глубокая умственная отсталость |
| F-78 | Другая умственная отсталость |
| F-79 | Неуточненная умственная отсталость |
Дата разработки/пересмотра протокола: 2015 год (пересмотрен в 2017 г.)
Сокращения, используемые в протоколе:
| АЛТ | - | аланинаминотрансфераза | |
| АСТ | - | аспартатаминотрансфераза | |
| ВВК | - | военно-врачебная комиссия | |
| ГОБМП | - | гарантированный объем бесплатной медицинской помощи | |
| DSM( Diagnostic and Statistical Manual of mental disorders) | - | диагностическое и статистическое руководство по психическим расстройствам | |
| КТ | - | компьютерная томография | |
| КП | - | клинический протокол | |
| ЛС | - | - лекарственные средства | |
| МЗСР | - | Министерство здравоохранения и социального развития | |
| МКБ | - | Международная классификация болезней | |
| МНН | - | международное непатентованное название (генерическое название) | |
| МРТ | - | магнитно-резонансная томография | |
| МСЭК | - | медико- социальная экспертная комиссия | |
| НПА | - | нормативно-правовой акт | |
| НЛФ | - | национальный лекарственный формуляр | |
| ОАК | - | общий анализ крови | |
| ОАМ | - | общий анализ мочи | |
| ПЭТ | - | позитронно-эмиссионная томография | |
| ПМПК | - | психолого-медико-педагогическая комиссия | |
| ППР | - | преждевременное половое развитие | |
| РЭГ | - | реоэнцефалография | |
| РК | - | Республика Казахстан | |
| У.о | - | Умственная отсталость | |
| ЭКГ | - | электрокардиограмма | |
| ЭПО | - | экспериментально-психологическое обследование | |
| ЭЭГ | - | электроэнцефалограмма | |
| ЭхоЭГ | - | эхоэлектроэнцефалограмма | |
Пользователи протокола: врачи общей практики, детские психиатры, психиатры.
Категория пациентов: дети, взрослые.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
| Этиопатогенетическая классификация для выделения этиологического фактора (по Г.Е.Сухаревой) | •У.о., вызванная поражением половых клеток (гаметопатии): болезнь Дауна, истинная микроцефалия, фенилкетонурия, галактозурия и др. энзимопатии; у.о., сочетающаяся с системными поражениями кожи и костной системы. |
| •У.о., связанная с эбриопатиями и фетопатиями: - связанная с внутриутробным инфекционным поражением (вирусы краснухи, гриппа, паротита, гепатита, цитомегалии, токсоплазмоза, сифилиса, листериоза и т.п.; - связанная с внутриутробным поражением экзо- и эндотоксическими агентами (при гормональных нарушениях у матери, при интоксикациях беременной); - обусловленная гемолитической болезнью новорожденного | |
| •У.о., связанная с интранатальными и постнатальными вредностями (родовая травма и асфиксия, ч.м.т. в раннем детстве до 3-х лет, нейроинфекции, перенесенные в раннем детстве | |
| Клинико-физиологическая классификация (по С.С.Мнухину, Д.Н. Исаеву) - для выделения клинических форм, для ориентации психиатров, дефектологов, логопедов, медицинских психологов на прицельную работу соответственно клиническим приоритетам, для составления индивидуальных абилитационных программ | •Легкая у.о. (легкая умственная субнормальность, дебильность, F-70); коэффициент умственного развития поWISC (стандартизированная методика Д.Векслера) - в диапазоне 50-69. •Умеренная у.о. (имбецильность легкой и средней степени, F-71); коэффициент умственного развития - 35-49. •Тяжелая у.о.(тяжелые варианты имбецильности, F-72); коэффициент умственного развития - 20-34. •Глубокая у.о.(идиотия, F-73 ); коэффициент умственного развития - ниже 20 баллов. |
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [3, 13-21]
Диагностические критерии [ 3, 13-21 ]
Жалобы:
· задержка или недоразвитие психических функций в виде выраженных эмоционально-волевых расстройств и моторики (запаздывание становления стато-моторных актов, отсутствие моторно-адаптивных движений);
· слабовыраженный интерес к окружающим;
· недоразвитие речи, процессов внимания и памяти, возможны судорожные приступы;
· поведенческие нарушения;
· задержка, либо неполное развитие психики, проявляющееся нарушением интеллекта и социальной дезадаптацией.
· существенное недоразвитие специфических для возраста видов деятельности - игры, рисования, конструирования, элементарного бытового труда.
Лабораторные исследования:
· исследование в соответствии с выявленными признаками (стигмы дизонтогенеза) на хромосомные и генные дефекты, сопряженные с умственной отсталостью.
· ОАК - лейкопения, лейкоцитоз, лимфоцитоз, анемия.
· ОАМ - лейкоцитоз, белок, эритроциты, бактерии.
· биохимический анализ крови - билирубин, АЛТ, АСТ;
· ПЦР - мазок из носа и горла на патогенную флору;
· ИФА на выявление вирусов кори, менингита, цитомегаловруса, герпеса и др., влияющих на формирование У.о.
Инструментальные исследования:
· ЭЭГмониторинг, ЭхоЭЭГ - при наличии в анамнезе судорожных состояний;
· R-графия черепа - при подозрении на токсоплазмоз; после перенесенных ч.м.т.; при микроцефалии и гидроцефалии и др. показаниях.
· КТ, ЯМРТ, ПЭТ - при подозрении на объемный процесс в головном мозге, при атрофии коры головного мозга и др. показаниям;
· УЗДГ сосудов головы и шеи, РЭГ - при признаках ликворной гипертензии, аневризмы сосудов головного мозга;
· аудиограмма - при подозрении на тугоухость или глухоту.
Показания для консультации специалистов:
· консультация генетика - верификация генной или хромосомной патологии;
· консультация детского невропатолога, невропатолога - для выявления органической патологии со стороны нервной системы, классификация пароксизмальных состояний, рекомендации по терапии антиконвульсантами;
· консультация логопеда - для диагностики уровня развития речи;
· консультация медицинского психолога (ЭПИ) - определение коэффициента умственной отсталости; выявление особенностей мыслительной деятельности (снижение уровня обобщения и конкретность мышления), исследование уровня развития эмоционально-волевой сферы и др., с помощью стандартизированных тестов в соответствии с возрастом ребенка;
· консультация дефектолога - определение уровня интеллектуального дефекта и разработка индивидуальной программы обучения;
· консультация сурдолога - исследование слуха;
· консультация эндокринолога - при избыточном весе, ППР;
· другие узкие специалисты - по показаниям.
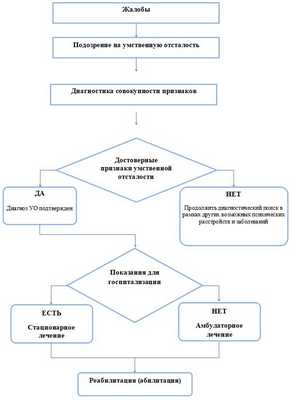
Диагностический алгоритм (схема)
Нейросенсорная тугоухость
Нейросенсорная тугоухость - нарушение слуха, обусловленное поражением слухового анализатора и проявляющееся односторонним или двусторонним снижением слуха, шумом в ушах, а также возникающими в связи с этим нарушениями социальной адаптации. Диагностика заболевания основана на изучении анамнеза, данных физикального и инструментального обследования (камертональных методов, аудиометрии, МРТ, УЗИ БЦА и др.). Лечение предусматривает восстановление сниженной слуховой функции при помощи слухопротезирования, использование глюкокортикоидов, медикаментозных средств с ангиопротекторным и нейропротекторным действием.
Общие сведения
Нейросенсорная тугоухость (НСТ, сенсоневральная глухота) - снижение функции слухового анализатора, проявляющееся частичной или полной потерей слуха. При этом патологический процесс может поражать структуры, участвующие в восприятии звука, на различных участках: в клетках внутреннего уха, в нервных проводниках, в стволе или коре головного мозга. По данным статистики, примерно 6% населения нашей планеты имеют нарушения слуха различной степени выраженности. Из них около 80-90% жалуются на шум в ушах. С возрастом нарушения слуха прогрессируют, от 30 до 60% людей старше 65-70 лет страдают тугоухостью.
Причины нейросенсорной тугоухости
Нейросенсорная потеря слуха может возникать в результате врожденных или приобретенных нарушений функции слуха.
1. Врожденная патология.
- пороки развития среднего или внутреннего уха, в том числе - обусловленные генетическими нарушениями (синдромы Ваарденбурга, Стиклера, Ушера, Пендреда, Ланге-Нильсена, Альпорта, нейрофиброматоз II типа, болезнь Рефсума);
- патология в родах (гипоксия плода).
2. Внешние факторы.
- инфекции (грипп, ОРЗ, паротит, корь, краснуха, скарлатина, менингит и др.);
- сосудистые расстройства при артериальной гипертензии, церебральном атеросклерозе;
- интоксикации (промышленные и бытовые токсины, медикаментозные средства с ототоксическим действием: аминогликозиды, антималярийные препараты, анальгетики, цитостатики и т. д.),
- травмы костей черепа;
- акустические повреждающие агенты и баротравма;
- эндокринные расстройства;
- болезни крови;
- неблагоприятные метеорологические факторы;
- физиологическое старение.
Вышеперечисленные внешние воздействия приводят к возникновению патологического процесса в слуховом анализаторе с развитием преходящей ишемии, стойкого нарушения кровообращения, а затем и гибели чувствительных клеток внутреннего уха, проводящего аппарата или корковых центров органа слуха.
Нейросенсорная тугоухость классифицируется по длительности и тяжести течения, уровню поражения, времени появления основной симптоматики и степени снижения слуха.
- Продолжительность. Симптомы НСТ могут появиться внезапно (за 3-6 часов, например, во время ночного сна) или постепенно (на протяжении 3-5 суток). Заболевание может приобретать хроническое течение со стабильным или прогрессирующим снижением слуха.
- Время появления. Ухудшение слуха может возникнуть в первые годы жизни ребенка, еще до развития полноценной речи (в долингвальный период), или уже после формирования речевой функции (постречевая тугоухость).
- Выраженность нарушений. Выделяют четыре степени тугоухости, которые определяют при сравнении с нормальными показателями. В норме слуховой порог находится в промежутке между 0 и 25 дБ, при первой степени НСТ он равен 26-30 дБ, при второй (умеренные нарушения) - 41-55, при третьей - 56-70, при четвертой (тяжелая степень) - 71-90 дБ. При полной глухоте этот показатель превышает 90 дБ.
Симптомы нейросенсорной тугоухости
Основными проявлениями заболевания являются снижение слуха и шум в ушах, реже наблюдается головокружение и сопутствующие соматоформные расстройства. Изменяется восприятие обычной разговорной и шепотной речи. При легкой степени НСТ обычный разговор слышен с расстояния 5-7 метров, а шепот - с 2-3 метров. При умеренных нарушениях эти показатели снижаются соответственно до 3-4 и 1 метра, при тяжелых разговорная речь слышна в лучшем случае с расстояния 1 метра, а шепот неразличим вообще. При IV степени нейросенсорной тугоухости человек неспособен воспринимать даже громкие звуки с самого близкого расстояния без специальных приборов.
Снижение слуха нередко сопровождается появлением шума в ушах периодического или постоянного характера. Шум может восприниматься в виде высокочастотных звуков по типу писка, звона, шипения, а также представлять собой постоянный надоедливый низкочастотный гул. При наличии сопутствующего кохлеовестибулярного синдрома больных беспокоят приступы головокружения, нередко сочетающиеся с тошнотой (иногда рвотой), признаки нарушения равновесия: ухудшается координация движений при выполнении простых бытовых манипуляций, появляется пошатывание при ходьбе, неустойчивость и высокая вероятность падения при резких поворотах.
Длительное хроническое течение нейросенсорной тугоухости со значительным нарушением слуховой функции становится причиной развития психоэмоциональных расстройств (снижение настроения, раздражительность, беспокойство, тревога), потере социальных контактов, снижению и утрате работоспособности (трудоспособности). В пожилом возрасте частичная или полная утрата слуха при отсутствии своевременной коррекции и наличии сопутствующих сосудистых заболеваний головного мозга нередко приводит к прогрессирующим нарушениям памяти, мышления, появлению бредовых и галлюцинаторных синдромов.
При остром развитии болезни клиническая симптоматика появляется внезапно (в течение 3-12 часов, часто во время ночного сна) на фоне полного благополучия. Иногда снижение слуха может быть более продолжительным (на протяжении 3-5 суток). При подостром и хроническом течении нейросенсорной тугоухости патологический процесс развивается в течение 1-3 месяцев и более.
Выявление этиологических факторов, определение выраженности нарушений слуха и наличия сопутствующих заболеваний, влияющих на течение НСТ, требует участия в диагностике врачей различных специальностей: отоларинголога, отоневролога, офтальмолога, кардиолога, эндокринолога, травматолога-ортопеда и других специалистов. Стандартное физикальное обследование, в частности, отоскопия, не дает какой-либо значимой информации, так как признаки поражения наружного уха и барабанной перепонки обычно отсутствуют. При этом простая оценка слышимости разговорной и шепотной речи на определенном расстоянии в кабинете ЛОР-врача позволяет ориентировочно оценить степень снижения слуха.
Более информативным является использование специальных инструментальных исследований: камертональных проб (Вебера, Ринне, Федеричи), тональной аудиометрии, регистрации слуховых потенциалов (электрокохлеографии), проведения вестибулометрических тестов. Для выявления сопутствующих заболеваний нервной системы и патологии позвоночника, исключения травматических повреждений может назначаться МРТ или КТ костей лицевого черепа и головного мозга, шейного отдела позвоночника, УЗИ брахиоцефальных артерий и т. д. Дифференциальная диагностика нейросенсорной тугоухости проводится с другими заболеваниями уха, горла и носа (хроническим средним отитом и связанными с ним кондуктивными нарушениями, болезнью Меньера, лабиринтитом, невриномой слухового нерва и др.), рассеянным склерозом, сосудистыми заболеваниями головного мозга (дисциркуляторной энцефалопатией, последствиями перенесенного инсульта, сосудистой деменцией).
Лечение нейросенсорной тугоухости
Основная цель лечебных мероприятий - восстановление или стабилизация функции слуха, устранение сопутствующей симптоматики (головокружения, шума в ушах, нарушений равновесия, нервно-психических расстройств), возвращение к активной жизни, социальным контактам.
- Физиотерапия, рефлексотерапия. На начальных стадиях заболевания применяется фоноэлектрофорез, электростимуляция тканей внутреннего уха, акупунктура и электропунктура, что позволяет в ряде случаев снизить интенсивность шума в ушах, избавиться от головокружения, улучшить сон и настроение.
- Медикаментозное лечение. Эффективность лекарственного воздействия наиболее высока при раннем начале лечения. При внезапно наступившей тугоухости полностью восстановить слух иногда позволяет применение ударных доз глюкокортикоидных гормонов в течение 5-8 суток. Широкое применение находят препараты, улучшающие кровообращение, проведение нервных импульсов и микроциркуляцию: пентоксифиллин, пирацетам. При сопутствующем НСТ головокружении назначают средства с гистаминоподобным действием, например, бетагистин. Используются медикаменты, оказывающие гипотензивное действие при наличии артериальной гипертензии, а также психотропные препараты при наличии нервно-психических расстройств.
- Слухопротезирование. Показано при умеренной и тяжелой степени утраты слуха. Применяются заушные, внутриушные и карманные аналоговые и цифровые аппараты для моноаурального или бинаурального слухопротезирования.
- Хирургическое лечение, кохлеарная имплантация. Практикуется транстимпанальное введение глюкокортикоидных гормонов в барабанную полость. Оперативные вмешательства проводятся при опухолях задней черепной ямки для уменьшения выраженности некоторых симптомов, сопровождающих вестибулярные расстройства. Кохлеарная имплантация выполняется при полном отсутствии слуха при условии сохранности функции слухового нерва.
Прогноз и профилактика
Прогноз у больных с острой нейросенсорной тугоухостью при своевременном лечении в 50% случаев относительно благоприятный. Применение слуховых аппаратов и имплантации при хронической НСТ обычно позволяет стабилизировать слух. Профилактические мероприятия по предотвращению утраты слуховой функции предусматривают исключение вредных факторов внешней среды (шума и вибрации на производстве и в быту), отказ от алкоголя и приема токсичных медикаментозных средств, предупреждение травматизма, в том числе акустических и баротравм, своевременное лечение инфекционных и соматических заболеваний.
Болезнь Рефсума ( наследственная полиневропатическая атаксия , полиневритоподобная гемератюпическая гередоатаксия , синдром Рефсума )
Болезнь Рефсума — генетически детерминированное расстройство окисления фитановой кислоты с ее накоплением в тканях организма, приводящим к неврологическим нарушениям, ухудшению зрения, слуха, обоняния, ихтиозным изменениям кожи, нарушениям со стороны сердца. Диагностируется по уровню фитановой кислоты в крови и моче, значительно превышающему норму. Дополнительно проводится исследование нервной системы, зрительной функции, слуха и обоняния, сердечной деятельности. Основу лечения составляет диета с ограничением нутриентов, содержащих фитановую кислоту. При тяжелом состоянии пациентам показан плазмаферез.
МКБ-10
Болезнь Рефсума — редкое заболевание, обусловленное генетически детерминированным дисметаболизмом фитановой кислоты с ее накоплением в различных тканях: в головном и спинном мозге, периферических нервных стволах, сердце, печени и почках. Болезнь Рефсума названа в честь норвежского невролога, впервые давшего описание патологии в 1946 г. В неврологии известна также как наследственная полиневропатическая атаксия, синдром Рефсума. Поскольку в патогенезе заболевания основным субстратом выступает дисметаболизм липидов, болезнь Рефсума можно отнести к наследственным липидозам, куда также относится болезнь Нимана-Пика, болезнь Тея-Сакса, болезнь Гоше и пр.
Возраст манифестации симптомов варьирует от 1 до 50 лет. В связи с таким широким диапазоном дебюта выделяют взрослую форму заболевания с началом после 10-летнего возраста и инфантильную — у детей 1-10 лет. Распространенность не установлена. Заболевают лица обоих полов.
Причины
Болезнь Рефсума характеризуется аутосомно-рецессивным путем наследования. На сегодняшний день выделены 2 генные мутации, обуславливающие заболевание: дефект гена фитаноил-КоА-гидроксилазы (10-я хромосома, локус 10pter-p11.2) и дефект гена пероксина 7 (6-я хромосома, локус 6q22-q24). Генетические нарушения детерминируют дефектное окисление фитановой кислоты и ее накопление в организме. В результате фитановая кислота замещает собой другие незаменимые жирные кислоты.
В первую очередь поражается нервная система. Дегенеративные изменения затрагивают периферические нервные стволы, передние рога и корешки спинного мозга, мозжечковые тракты, зрительные, глазодвигательные, обонятельные и слуховые нервы. Поражается сетчатка глаз с развитием пигментной ретинопатии. Накопление фитановой кислоты в миокарде приводит к формированию кардиомиопатии. Дегенеративные изменения в нервах проводящей системы сердца обуславливают возникновение аритмии. Замещение линолевых и арахидоновых кислот дермы на фитановую кислоту приводит к ихтиозу.
Симптомы болезни Рефсума
Множественные поражения периферических нервных стволов проявляются симптомами полиневропатии: онемением кистей и стоп, парестезиями, снижением болевого восприятия и глубокой (проприоцептивной, вибрационной) чувствительности. Полиневропатия носит сенсомоторный характер. Двигательные нарушения начинаются преимущественно с пареза мышц-разгибателей стоп, в дальнейшем вялые парезы распространяются на дистальные отделы ног и рук, сопровождаются гипотрофиями мышц стоп, голеней, кистей и предплечий. Характерна мозжечковая атаксия с шаткостью ходьбы, гиперметрией, изменением почерка. Со стороны зрения наблюдается снижение его остроты, гемералопия, сужение зрительных полей, в ряде случаев — катаракта. Иногда имеет место фотофобия.
Зачастую болезнь Рефсума сопровождается тугоухостью, утратой остроты обоняния (аносмия), опущением верхнего века, глазодвигательными расстройствами. Отмечается сухость кожи и шелушение. Степень проявленности ихтиозных изменений весьма вариабельна: от легкого гиперкератоза стоп и ладоней с образованием мелких белых корочек до тяжелого пластинчатого ихтиоза. У некоторых пациентов имеются сопутствующие скелетные дисплазии: искривление позвоночника (сколиоз), деформации стоп (полая стопа, искривление и укорочение пальцев стопы).
Подтвердить диагноз позволяет анализ крови и мочи с исследованием концентрации фитановой кислоты. При болезни Рефсума этот показатель доходит до 800 мкмоль/л, в то время как в норме он не превышает 19 мкмоль/л. Всем пациентам проводится консультация офтальмолога. Проверка остроты зрения выявляет ее снижение, периметрия устанавливает концентрическое сужение зрительных полей, при офтальмоскопии обнаруживаются признаки атрофии зрительных нервов, полиморфные дегенеративные изменения сетчатки.
Электронейромиография позволяет определить полиневропатический характер поражения периферической нервной системы, выявить патологию мотонейронов передних рогов. При исследовании цереброспинальной жидкости диагностируется повышенный уровень альбуминов. Расстройство обоняния выявляется путем ольфактометрии. При нарушениях слуха показана консультация сурдолога и аудиометрия.
Наличие костных деформаций является поводом для назначения рентгенографии позвоночника, КТ или рентгенографии стоп с последующей консультацией ортопеда. Для оценки состояния сердца выполняется ЭКГ, пациентов консультирует кардиолог. С целью определения типа наследования патологии проводится генеалогическое исследование. Окончательный диагноз может быть подтвержден путем ДНК-анализа.
В ходе диагностики болезнь Рефсума дифференцируют от амиотрофии Шарко-Мари-Тута, полиневропатий другого генеза (синдрома Дежерина-Сотта, диабетической нейропатии, синдрома Русси-Леви, невропатии при паранеопластическом синдроме и др.), атаксии Фридрейха.
Лечение болезни Рефсума
Фитановая кислота не образуется в организме, поэтому ограничение ее экзогенного поступления является эффективным способом лечения. Диета заключается в ограничении поступления фитановой кислоты до 5 мг в сутки. Именно такое количество фитановой кислоты организм больного способен метаболизировать, благодаря имеющимся альтернативным механизмам ее окисления. Из рациона рекомендуется исключить употребление зеленых овощей, зелени и животных жиров. Во избежание быстрой потери веса диета должна включать большое количество углеводов. Строгое соблюдение диеты быстро приводит к улучшению клинического статуса пациентов. Уменьшается выраженность полиневропатии и парезов, нормализуется координация, улучшается состояние кожи. Постоянно придерживаться диеты больным необходимо пожизненно.
В тяжелых случаях для очистки организма от избыточного количества фитановой кислоты применяется плазмаферез. С целью уменьшения неврологических симптомов назначаются антихолинэстеразные фармпрепараты (галантамин, неостигмин), витамины группы В, лечебная физкультура, массаж. При деформации стоп и/или парезе мышц-разгибателей рекомендовано ношение ортопедической обуви. Выраженная двусторонняя тугоухость является показанием к проведению кохлеарной имплантации.
Прогноз
Прогноз при правильном лечении благоприятный. При отсутствии терапии заболевание продолжает медленно прогрессировать, приводя к выраженным парезам и атаксии, значительному падению зрения и слуха, нарушениям сердечной деятельности. Последнее может стать причиной внезапной смерти пациента.
Синдром Жубер
Синдром Жубер - редкое генетически гетерогенное наследственное заболевание, характеризующееся нарушением формирования мозжечка и структур мозгового ствола с развитием соответствующей неврологической симптоматики. Симптомы данной патологии проявляют значительную вариабельность по своей выраженности, наиболее часто наблюдаются расстройства дыхания, глазодвигательные нарушения и мышечная слабость, возможны нарушения слуха и отставание в интеллектуальном развитии. Диагностика синдрома Жубер производится на основании неврологического осмотра больного, магнитно-резонансной томографии головного мозга, молекулярно-генетических исследований. Специфического лечения на сегодняшний момент не существует, применяют поддерживающие и симптоматические мероприятия.
Синдром Жубер (Жуберт) - достаточно редкая генетическая патология, которая характеризуется нарушением эмбрионального развития важных структур головного мозга, что приводит к различным неврологическим проблемам. Впервые данное заболевание было описано канадским педиатром Мари Жуберт в 1969 году. Она выявила у четырех детей, чьи родители состояли между собой в кровном родстве, нарушения дыхания, слуха, мышечную слабость и признаки умственной отсталости. С 1977 года подобные нарушения выделили в отдельную нозологическую единицу под названием «синдром Жубер». В дальнейшем врачи-генетики смогли определить значительную генетическую гетерогенность заболевания - на сегодняшний момент известно не менее 20 генов, мутации которых смогли связать с этой патологией. При этом почти в половине случаев синдрома Жубер его молекулярно-генетические механизмы остаются неизвестными. По последним данным, встречаемость этого заболевания составляет примерно 1 случай на 1 млн. новорожденных, механизм передачи аутосомно-рецессивный, мальчики и девочки поражаются с одинаковой частотой. Каких-либо национальных, расовых или региональных особенностей в распределении синдрома Жубер не выявлено.
Причины и классификация синдрома Жубер
По мере изучения синдрома Жубер обнаруживается все больше генов, мутации которых способны приводить к развитию этого заболевания. На сегодняшний день таких генов выявлено почти два десятка, при этом примерно в половине клинических случаев синдрома Жубер определить генетические нарушения не удается, что говорит о роли других, еще не изученных дефектов. Мутации генов, приводящие к этому заболеванию, возникают на различных хромосомах, но всегда наследуются по аутосомно-рецессивному механизму. Также все эти гены объединяет участие в эмбриональном развитии структур головного мозга, именно это обстоятельство служит причиной появления характерной симптоматики синдрома Жубер.
Установлено, что мутации одних генов приводят к развитию заболевания несколько чаще, чем другие. Одним из них является ген AHI1, который располагается на 6-й хромосоме. Согласно данным медицинской статистики, дефекты этого гена становятся причиной 10-12% всех случаев синдрома Жубер. Ген AHI1 кодирует специфический белок, принимающий активное участие в формировании некоторых элементов ствола мозга и сетчатки глаза. Другой распространенной причиной синдрома Жубер считается дефект гена CEP290, локализованного на 12-й хромосоме. Он кодирует последовательность белка, принимающего участие в формировании клеточных центросом и ресничек в различных органах организма (головной мозг, сетчатка глаза, сердце, легкие, почки). Мутации гена CEP290 выявляются примерно у 10% больных синдромом Жубер.
Кроме того, к синдрому Жубер могут приводить мутации таких генов, как TCTN1 и TCTN2 (12-я хромосома), TMEM138 и TMEM216 (11-я хромосома), TMEM237, TTC21B и NPHP1 (2-я хромосома), ARL13B (5-я хромосома) и целый ряд других. Они встречаются с частотой в несколько процентов от всех случаев заболевания, для многих относительная распространенность неизвестна. Те гены, функции которых удалось установить, также контролируют развитие ресничек, центриолей или цитоскелета, что позволяет считать синдром Жубер проявлением нарушений формирования именно этих структур. Мутации всех вышеперечисленных генов наследуются по аутосомно-рецессивному механизму, однако в последние годы появились указания на возможность сцепленной с полом передачи. Предполагают, что такая форма синдрома Жубер обусловлена мутацией гена OFD1, локализованного на Х-хромосоме.
Симптомы синдрома Жубер
Фенотипические проявления синдрома Жубер в целом сходны при различных генетических разновидностях заболевания. Вместе с тем, имеются незначительные различия. Выраженность симптоматики может значительно различаться даже в пределах одной семьи. В настоящее время достоверно неизвестны причины того, почему тяжесть течения синдрома Жубер отличается у разных больных. В подавляющем большинстве случаев заподозрить наличие заболевания можно в первые дни жизни ребенка - выявляется мышечная гипотония, аномальные движения глаз, часто определяется колобома (дефект оболочек глаза). Характерным признаком синдрома Жубер являются нарушения дыхания - нестабильность ритма (тахипноэ, брадипноэ), возможна остановка дыхания во время сна (ночное апноэ).
По мере роста ребенка отмечается незначительное прогрессирование заболевания - мышечная гипотония перетекает в мозжечковую атаксию, наблюдается отставание в моторном и интеллектуальном развитии. При этом спектр нарушений интеллекта при синдроме Жубер колеблется в очень широких пределах - от нормы до глубокой умственной отсталости. Возможны нарушения слуха (вплоть до нейросенсорной глухоты) и зрения (обусловленные как глазодвигательными аномалиями, так и дистрофией сетчатки). Примерно у половины больных синдромом Жубер развиваются различные аномалии внутренних органов - фиброз печени, поликистоз почек, врожденные пороки сердца. Реже возникает энцефалоцеле (через большое затылочное отверстие или дефекты свода черепа), гидроцефалия, гамартомы полости рта.
Из-за значительного диапазона выраженности проявлений синдрома Жубер риски летального исхода и различных осложнений достаточно неопределенные. В самых тяжелых случаях возможна смерть больных в младенческом возрасте из-за дыхательных и неврологических нарушений. При менее тяжелом течении синдрома Жубер пациенты могут доживать до взрослого и даже преклонного возраста, при этом они, как правило, лишены возможности ходить из-за мозжечковой атаксии. Основные риски в подобных случаях создают поражения и аномалии развития внутренних органов - сердца, почек, печени.
Диагностика и лечение синдрома Жубер
Для определения синдрома Жубер применяют следующие диагностические методики: неврологический осмотр больных, магнитно-резонансную томографию головного мозга, дополнительные исследования глаз, слуха и работы внутренних органов. Молекулярно-генетическая диагностика в большинстве современных лабораторий возможна в отношении четырех наиболее распространенных типов заболевания - обусловленных мутациями генов AHI1, CEP290, CC2D2A и TMEM67. При осмотре больных синдромом Жубер определяется мышечная слабость, признаки мозжечковой атаксии и нарушений координации, основные сухожильные рефлексы резко снижены. Практически всегда обнаруживается отставание в моторном развитии, в ряде случаев - различная степень умственной отсталости.
Самым типичным диагностическим признаком синдрома Жубер является наличие так называемого «симптома молярного зуба» - характерные изменения на МРТ головного мозга, внешне похожие на разрез зуба. Это проявление говорит о наличии нарушений формирования стволовых элементов мозга. Также на магнитно-резонансной томографии часто определяется недоразвитие червя мозжечка, гипоплазия мозолистого тела, гидроцефалия, расширение желудочков, энцефалоцеле и другие аномалии развития головного мозга. У взрослых больных синдромом Жубер нередко выявляются признаки поражения внутренних органов - поликистоз почек, фиброз печени, нарушения сердечного ритма. При осмотре у офтальмолога часто обнаруживаются непроизвольные аномальные движения глаз (нистагм), колобома, дистрофия и дегенерация сетчатки.
При помощи методов современной генетики возможна молекулярно-генетическая диагностика синдрома Жубер, который вызывается мутациями генов AHI1, CEP290, CC2D2A и TMEM67. В общей сложности дефекты этих генов обуславливают порядка 40% всех случаев заболевания. По этой причине отрицательный результат генетических анализов не является поводом для гарантированного исключения синдрома Жубер. Вспомогательную роль в определении патологии играет изучение наследственного анамнеза больного с целью подтверждения аутосомно-рецессивной передачи. При помощи прямого автоматического секвенирования можно выявлять носительство патологической формы гена у родственников больного или при отягощенной по этому состоянию наследственности.
Специфического лечения синдрома Жубер не существует, медицинская помощь при этом заболевании сводится к паллиативным и симптоматическим мероприятиям. Для ослабления неврологических симптомов применяют ноотропные средства - их регулярный прием, начатый с раннего возраста, может значительно улучшить прогноз в отношении интеллектуального развития больного. Также при синдроме Жубер используют различные методы физиотерапии, специальные упражнения для улучшения координации движений и уменьшения проявлений атаксии. В раннем возрасте часто необходим контроль дыхания больного во избежание потенциально опасного апноэ.
Прогноз и профилактика синдрома Жубер
Прогноз синдрома Жубер часто неопределенный, поскольку находится в зависимости от выраженности симптомов и тяжести клинического течения заболевания. В самых тяжелых случаях возможен летальный исход еще в раннем детстве из-за нарушений дыхания и неврологических патологий. В большинстве случаев больные доживают до взрослого и даже пожилого возраста, хотя совокупность атаксии, патологий внутренних органов, нарушений зрения и слуха часто приводит к глубокой инвалидизации. Умственное развитие может быть сохранено, но встречаются и различные степени умственной отсталости. Профилактика синдрома Жубер возможна только в качестве пренатальной диагностики заболевания и определения носительства патологической формы гена у лиц с отягощенной наследственностью.
Глухота с изменениями сетчатки, мышечной слабостью и умственной отсталостью
ЛОР-болезни:
Популярные разделы сайта:
Синдромы Рефсума, Маршалла. Сочетание миопии с нарушением слуха
При синдроме Рефсума [Refsum] — наследственной атаксической полииефритоформной нейропатии — пигментный ретинит и нейросенсорпая тугоухость встречаются в половине случаев. Синдром передается по рецессивному типу и характеризуется сужением полей зрения, ночной слепотой, гипертрофической периферической нейропатией, мозжечковой атаксией, нистагмом, увеличением содержания в крови и мозге липидных субстанций. В связи с этим рекомендуются диета с низким содержанием фитановой кислоты и назначение фитола.
Кроме пигментного ретинита, нейросенсорная тугоухость может сочетаться с гипогонадизмом [Reinstein, Chalfin]. С детского возраста прогрессирует потеря слуха на звуки частотой свыше 2000 Гц. Заболевание наследуется по аутосомно-рецессивному типу.
Нейросенсорная тугоухость сопровождает синдром Маршалла [Marshall], включающий симптомокомплекс в виде седловидного носа, катаракты (врожденной или юношеской), миопии и скелетных аномалий. Синдром передается по аутосомно-доминантному типу.
Миопия, голубые склеры, иногда кератоконус и морфаноподобный вид больного описаны Walker как обязательные симптомы. Прогрессирующая нейросенсорная тугоухость в диапазоне высоких частот начинается в возрасте 7—9 лет. Предполагается аутосомно-рсцессивное наследование этого синдрома. При дифференциальной диагностике необходимо исключить синдром Элерса—Данло и синдром кератоконуса, голубых склер, слабости связок и коидуктивной глухоты.
Нейросенсорная тугоухость может быть спутником и других форм близорукости. В 1965 г. Flyun и Aird сообщили о 15 больных из одной семьи, у которых синдром включал миопию, катаракту, пигментный ретинит, периферическую миопатию с. приступами болей, потерей чувствительности и мышечной миотрофией, скелетные аномалии (в частности, кифосколеоз), судорожные припадки, изменения ЭЭГ и иейросенсорную глухоту. Передача этого синдрома осуществляется по аутосомно-доминантному типу. Врожденная нейросенсорная тугоухость с миопией и умственной отсталостью описаны Eldridge и соавт. в 1968 г. При данной патологии возможны протеинурия и гематурия. Этот синдром необходимо дифференцировать от синдрома Альпорта. Легкая степень миопии отмечена Sturtz и Burke. Различия заключаются в типе наследования: синдром Альпорта наследуется по доминантному типу.
Другое сочетание нейросенсорной тугоухости с близорукостью выявлено Holmes Schepens. Это заболевание наследовалось по аутосомно-рецессивному типу, характеризовалось вторичным телекантусом (гипертелоризм), выступающими бровями, атрофией хориоидеи, катарактой, гипоплазией стромы радужной оболочки глаз, возможной отслойкой сетчатки. Лабораторно определяется гипераминоацидурия.
Нейросенсорная тугоухость отмечается при синдроме Розенберга—Чуториана [Rosenberg, Chutcrian], для которого характерны атрофия зрительных нервов и полинейропатия. Наследование не установлено, но, по-видимому, по Х-сцепленному или аутосомно-рецессивному типу. Прогрессирующая тугоухость развивается в возрасте 5—6 лет. Ухудшение зрения до слепоты и нейросенсорная тугоухость в возрасте до 10 лет могут сопровождаться сахарным диабетом с легким течением. Передача этого синдрома аутосомно-рецессивная [Stevens, Macfayden]. Доминантный тип наследования прогрессирующей атрофии зрительных нервов и мейросенсорной тугоухости наблюдали Konigsmark и соавт..
Silvester в 1958 г. сообщил о нейросенсорной тугоухости, сочетающейся с атрофией зрительных нервов и атаксией, которая наследуется по доминантному типу, но отличается от атаксии Фридрейха.
Нейросенсорная тугоухость может наблюдаться при дисплазии или дистрофии роговицы. Например, синдром Харбойяна [Harbojan et al.,] может сочетаться с нарушением обмена кальция. Нейросенсорная тугоухость отмечается при синдроме Норри — помутнении хрусталика с пролиферацией ретролентальных масс [Norrie].
Своеобразный синдром «пигментного ретинита, наружной офтальмоплегии и полной сердечной блокады» описан в 1958 г. Kearns и Sayre. Этот синдром может быть неполным (без кардномиопатии). У больных отмечаются изменения по типу синдрома Морганьи — Адамса — Стокса.
Таким образом, изменения органа зрения могут быть определяющими при обосновании аудиологических исследований. С другой стороны, изменения слуха и особенности семейного анамнеза могут быть использованы для прогнозирования поражения органа зрения и ЦНС.
Читайте также: