Классификация спинальной мышечной атрофии (СМА) у детей
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Спинальные мышечные атрофии - группа клинически и генетически гетерогенных наследственных заболеваний, вызванных прогрессирующей дегенерацией мотонейронов передних рогов спинного мозга (1). Начало заболевания варьирует от рождения до взрослого возраста. Является орфанным заболеванием (2)
Этиология и патогенез: Генетическое заболевание, при котором возможны все типы наследования (аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный). Ген SMN ответственен за развитие спинальной мышечной атрофии детского возраста с аутосомно-рецессивным типом наследования (1,6)
Название протокола: Спинальные мышечные атрофии у детей
Код(ы) по МКБ-10:
| МКБ-10 | |
| Код | Название |
| G 12 | Спинальная мышечная атрофия и родственные синдромы |
| G 12.0 | Детская спинальная мышечная атрофия, I тип (Верднига-Гоффманна) |
| G 12.1 | Другие наследственные спинальные мышечные атрофии |
| G 12.2 | Болезнь двигательного нейрона |
| G 12.8 | Другие спинальные мышечные атрофии и родственные синдромы |
| G 12.9 | Спинальная мышечная атрофия неуточненная |
Дата разработки/пересмотра протокола: 2018 год.
Сокращения, используемые в протоколе:
| АД | аутосомно-доминантный |
| АР | аутосомно-рецессивный |
| ГЭФР | гастроэзофагеальный рефлюкс |
| ЖКТ | желудочно-кишечный тракт |
| ИВЛ | инвазивная вентиляция легких |
| КТ | компьютерная томография |
| КФК | креатинфосфокиназа |
| МРТ | Магниторезонансная томография |
| НИВЛ | неинвазивная вентиляция легких |
| ПЦР | полимеразная цепная реакция |
| РКИ | рандомизированное клиническое исследование |
| СМА, SMA | спинальная мышечная атрофия |
| УЗИ | ультразвуковое исследование |
| ЭМГ | электромиография |
| MLPA | multiplex dependent probe amplification |
| SMN | сокр. от Survival Motor Neuron (ген «выживаемости мотонейрона» - англ.) |
Пользователи протокола: детские неврологи, неонатологи, педиатры, врачи общей практики, реабилитологи, детские хирурги-ортопеды, анестезиологи-реаниматологи, пульмонологи, клинические генетики.
Категория пациентов: дети.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортных или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты, которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GCP | Наилучшая клиническая практика. |

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
- Спинальная амиотрофия I типа (спинальная амиотрофия раннего детского возраста, или болезнь Верднига-Гоффмана).
- Спинальная амиотрофия II типа (спинальная амитрофия детского возраста, промежуточная форма).
- Спинальная амитрофия III типа (болезнь Кугельберга-Веландера).
- Бульбоспинальная амиотрофия Кеннеди (форма взрослых).
- Дистальная.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ (1,3,4,5-14)
Диагностические критерии
Жалобы: практические отсутствие или позднее формирование двигательных навыков.
- СМА 1 типа характеризуется дебютом ранее 6 месяцев, симптомокомплексом «вялого» ребенка, колоколообразной формой грудной клетки, выраженной гипотонией, арефлексией, фасцикуляциями языка и проблемами с дыханием. Пациенты обычно погибают до 2 лет от дыхательной недостаточности, возникающей в результате интеркурентных инфекций.
- СМА II типа характеризуется началом заболевания в возрасте от 6 мес. до 1,5 года и более медленным прогрессированием, тип наследования аутосомно-рецессивный. У больных также отмечается симптомокомплекс «вялого» ребенка, гипотония, арефлексия, фасцикуляции языка и проблемы с дыханием. Такие пациенты максимально способны самостоятельно сидеть, и у них развиваются многочисленные контрактуры крупных суставов
- СМА III типа развивается в возрасте от 1,5 лет, в большинстве случаев прогрессирует медленно, тип наследования аутосомно-рецессивный. эти пациенты могут самостоятельно ходить. Пациенты обычно имеют слабость в подвздошных, четырехглавых и аддукторных мышцах, гипотонию, гипорефлексию и фасцикуляции языка. Некоторые из пациентов этой группы со временем утрачивают способность к самостоятельному передвижению.
- СМА IV типа (бульбоспинальная амиотрофия Кеннеди, форма взрослых) проявляется в среднем на 4 десятилетии жизни со слабости бульбарных мышц (дисфагия, дизартрия) с последующим присоединением слабости проксимальных отделов конечностей, слабость мимической мускулатуры, атрофии и фасцикуляции в языке, генерализованные фасцикулякуции, крампи, постуральный тремор, сенсорная полиневропатия. Часто характерны гинекомастия, снижение половой функции, гипогонадизм, нарушение сперматогенеза, бесплодие, сахарный диабет. Тип наследования - Х-сцепленный рецессивный.
- Изменения нервно-мышечной системы: фибрилляции мышц языка, генерализованная мышечная гипотония, атрофии мышц и фасцикуллярные подергивания в мышцах спины, туловища, проксимальных (реже в дистальных) отделах верхних и нижних конечностей; гипорефлексия вплоть до арефлексии;
- Изменения костно-суставной системы: деформация грудной клетки, деформация позвоночника (кифосколиоз), контрактуры суставов, патология стоп.
- Нарушение дыхательной функции: как результат нарушения откашливания и глотания, гиповентиляции во время ночного сна, недоразвития\деформации грудной клетки, частных инфекционных заболеваний вследствие нарушения эвакуации секреторного отделяемого из дыхательных путей.
- Дисфункция ЖКТ: нарушения глотания (в т.ч. из-за бульбарного синдрома) нарушения моторики ЖКТ, которые включают запоры, задержку эвакуации содержимого желудка и потенциально опасный для жизни гастроэзофагальный рефлюкс (ГЭФР).
- Болевой синдром: как последствие патологии опорно-двигательного аппарата, остеопении и переломов.
- Нарушение роста и гипо-/гипертрофия.
- Нарушения чувствительности: не характерно.
- Нарушения психоречевого и когнитивного развития: не характерно
- Дети, которые не могут сидеть без посторонней помощи («лежачие пациенты»);
- Дети, которые могут самостоятельно сидеть, но не могут ходить без посторонней помощи («сидячие пациенты»);
- Дети, которые могут самостоятельно ходить («ходячие пациенты»).
- Общий анализ крови и мочи: специфических изменений нет.
- Биохимический анализ крови: уровень КФК может быть нормальным или слегка повышенным. Интерпретация для оценки задержки двигательного аппарата:
- в некоторых случаях целесообразна ликворограмма - повышение количества белка на 25 % и более.
- Электромиография (ЭМГ): ритмичные потенциалы фасцикуляций с амплитудой до 300 мкВ и частотой 5-35 Гц (ритм частокола). Скорости проведения импульса по периферическим двигательным волокнам могут быть как нормальными, так и немного сниженными за счет вторичных денервационных изменений. Для детей 1 и 2 типов имеет меньшее диагностическое значение, чем для иных типов.
- УЗИ и МРТ мышц: признаки жирового замещения мышечной ткани.
- Генетические тесты - предназначены для установления диагноза СМА, прогнозирования и выбора терапевтических подходов. Отсутствие полных копий SMN1 подтверждает диагноз, информация о копиях SMN2 является важной для прогноза и выбора терапии.
- Количественная ПЦР: идентифицирует гомозиготную делецию SMN1, но не позволяет подсчитать количество копий SMN1 и SMN2.
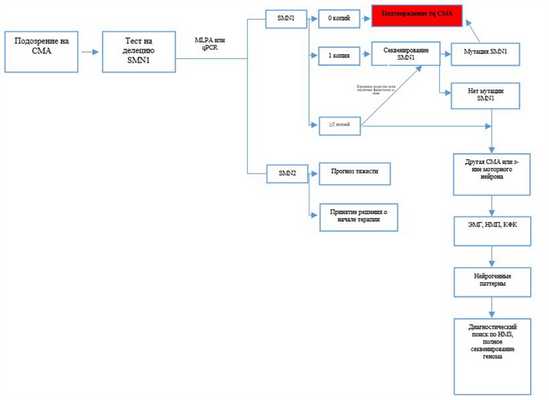
Диагностический алгоритм:
Примечания: ЭМГ - электромиография, НМП- нервно-мышечная проводимость; КФК - креатинфосфокиназа; НМЗ- нервно-мышечные заболевания
Зависимость клинической картины СМА от количества копий генов SMN1 и SMN2.
| Тип СМА | Количество копий генов | Особенности клинической картины |
| СМА | Полное отсутствие обоих генов SMN1 и SMN2 | Летальная ситуация |
| СMA тип 0 | Нет SMN1 гена и 1 копия SMN2 гена. | Тяжелая мышечная слабость, смерть наступает до 1 месяца |
| СMA тип I | Преимущественно делеции SMN1 или несколько миссенс мутаций в SMN1; SMN2 обычно 2 копии. | Симптомокомплекс «вялого» ребенка, смерть наступает до 2 лет. |
| СMA тип II | Мутации превращают ген SMN1 в SMN2; Копий гена SMN2 > 3 копий; Могут встречаться миссенс точечные мутации. | Могут самостоятельно сидеть. |
| СMA тип III | Копий гена SMN2 > 3 копий; Могут встречаться миссенс точечные мутации. | Могут самостоятельно ходить. |
| Специалист | Цель |
| Детский невролог | Клиническая диагностика заболевания, направление на генетическую и параклиническую диагностику. Разработка краткосрочного и долгосрочного плана ведения и реабилитации. Координатор мультидисициплинарной команды специалистов, мониторинг и оценка эффективности комплексного плана лечения и реабилитации. Принятие решения о назначении специфической терапии. |
| Генетик | Генетическая верификация диагноза. Медико-генетическое консультирование семьи, информирование о методах пренатальной и предимплатанционной диагностики. |
| Педиатр | Диагностика и коррекция нарушений со стороны внтуренних органов. Мониторинг физического, соматического и нутритивного статуса. |
| Пульмонолог/специалист по респираторной поддержке | Диагностика нарушений дыхательной системы, разработка и реализация плана лечения и долгосрочной курации в случае их наличия |
| Анестезиолог-реаниматолог (детский) | Диагностика нарушений дыхательной системы пациентов, нуждающихся в проведении неинвазивной вентиляции легких (НИВЛ), коррекция водно-электролитного обмена и белкового статуса на фоне дефицита массы тела тяжелой степени. |
| Гастроэнтеролог | Диагностика и коррекция нарушений пищеварительной системы, разработка и реализация плана лечения и долгосрочной курации в случае их наличия. |
| Диетолог | Решение вопросов подбора и реализации диеты |
| Ортопед | Диагностика нарушений костно-суставной системы, консервативная коррекция патологии позвоночника, суставов, стоп; хирургическая коррекция. Подбор ортезов/туторов и иных необходимых приспособлений. |
| Реабилитолог (в т.ч. специалист ЛФК) | Разработка и реализация комплексной реабилитации (в т.ч. двигательной). Обучение семьи пациента. |
| Психолог | Квалификация психологических нарушений, разработка и реализация плана лечения и долгосрочной курации в случае их наличия. Семейное психологическое консультирование |
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований
Дифференциальная диагностика СМА и других поражений спинного мозга
| Диагноз Признак | Боковой амиотрофический склероз | Вертеброгенная шейная миелопатия | Сирингомиелия |
| Клиника | Периферические парезы, высокие сухожильные периостальные рефлексы, фибрилляции, фасцикулляции мышц, поражение черепно-мозговых нервов (V-XII), атрофии | Периферические парезы и расстройства чувствительности в зонах иннервации шейных сегментов, доминируют симптомы ишемического поражения двигательных структур шейного утолщения | Дистальные атрофии, мышечный тонус и рефлексы снижены, болевой синдром, расстройства чувствительности диссоциированного типа, вазомоторные и трофические нарушения, дизрафический статус |
| Течение | Прогредиентное | Медленно прогрессирующее | Медленно прогрессирующее |
| Рентгенография | Без особенностей | Шейного отдела - выраженные явления остеохондроза и сужение позвоночного канала | Кифосколиоз, добавочные ребра, не заращение дужек шейных и поясничных позвонков, ассимиляция атланта с затылочной костью, базилярная импрессия |
Другие редкие формы спинальных мышечных атрофий представлены в таблице. Некоторые из них имеют единичные описания в мировой литературе, многие из которых специфичны для конкретных изолятов.
Дифференциальная диагностика проксимальных аутосомно-рецессивных СМА и других форм СМА
Спинальные амиотрофии ( Спинальные мышечные атрофии )
Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.
Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.
Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
- бульбоспинальная амиотрофия Кеннеди;
- скапулоперонеальная;
- лицелопаточноплечевая и окулофарингеальная формы;
- дистальная СМА;
- мономелическая СМА.
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Болезнь Верднига-Гоффмана
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Амиотрофии имеют восходящий характер — вначале возникают в ногах, затем вовлекаются руки, позже — дыхательная мускулатура, мышцы глотки и гортани. Сопровождается задержкой психического развития. К 1,5 годам наступает смертельный исход.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Ювенильная спинальная амиотрофия Кугельберга-Веландера
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Бульбоспинальная амиотрофия Кеннеди
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Дистальная СМА Дюшенна-Арана
Может иметь как рецессивный, так и доминантный тип наследования. Дебют приходится чаще на 20-летний возраст, но может произойти в любой период до 50 лет. Амиотрофии начинаются в кистях рук и приводят к формированию «когтистой кисти», затем охватывают предплечье и плечо, в связи с чем рука приобретает вид «руки скелета». Парезы мышц голеней, бедер и туловища присоединяются гораздо позже. Описаны случаи манифестации заболевания монопарезом (поражением одной руки). Прогноз благоприятный, за исключением случаев сочетания данного вида СМА с торсионной дистонией и паркинсонизмом.
Скапуло-перонеальная СМА Вюльпиана
Манифестирует в период от 20 до 40 лет амиотрофиями плечевого пояса. Типичны «крыловидные лопатки». Затем присоединяется поражение перонеальной группы мышц (разгибатели стопы и голени). В ряде случаев вначале поражаются перонеальные мышцы, а затем плечевой пояс. Спинальная амиотрофия Вюльпиана отличается медленным течением с сохранностью способности передвигаться спустя 30-40 лет от ее дебюта.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем - эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте - педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов - эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера - после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
- Лабораторные исследования. В лабораторных анализах практически все показатели находятся в пределах нормальных значений. Исключение составляет концентрация креатинфосфокиназы, которая у небольшой части пациентов может быть высокой.
- ЭМГ. При проведении игольчатой электромиографии обнаруживаются признаки дегенерации двигательных спинномозговых нейронов - снижение скорости и амплитуды вызванных потенциалов действия, регистрация спонтанной биоэлектрической активности в покое (фасцикуляций, фибрилляций), «ритм частокола».
- Биопсия мышц. При гистологическом исследовании мышечной ткани отмечаются типичные для амиотрофии изменения: некроз миофибрилл, разрастание жировой и соединительной ткани, чередование атрофии и гипертрофии в сочетании с интактными участками мышечной ткани.
- Спирометрия. При поражении дыхательной мускулатуры во время выполнения функции внешнего дыхания выявляются рестриктивные нарушения в виде снижения жизненной емкости легких.
- Генетический анализ. Основной метод диагностики, позволяющий достоверно установить диагноз спинальной амиотрофии. С помощью полимеразной цепной реакции обнаруживаются генетические мутации.
Дифференциальная диагностика
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
- псевдогипертрофическую мышечную дистрофию Дюшенна;
- ювенильный боковой амиотрофический склероз;
- детский церебральный паралич.
Лечение спинальных амиотрофий
Немедикаментозная терапия
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
- Обеспечение питания. При значительном нарушении акта глотания особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение пациента вертикальным. Может понадобиться установка назогастрального зонда.
- ЛФК. С целью повышения тонуса мышц и замедления их атрофии рекомендуются регулярные физические нагрузки. Наиболее эффективно совмещение активных (выполняются специалистом) и пассивных упражнений (выполняются самим пациентом).
- Физиолечение. Для активации метаболизма в мышечных тканях назначаются сеансы электростимуляции модулированным током, грязевые аппликации, электрофорез.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах выполняются различные виды массажа - ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Ортопедическое лечение. Для предупреждения и коррекции костных деформаций и суставных контрактур рекомендуется использование ортопедических приспособлений: корсетов, ортезов, ортопедической обуви.
- Респираторная поддержка. Нередко возникает необходимость в устранении кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Медикаментозная терапия
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
- Метаболические средства. Для улучшения обменных процессов в мотонейронах и мышечных тканях применяются препараты, стимулирующие функцию митохондрий (коэнзим Q10, янтарная кислота), ноотропы (пирацетам, гамма-аминомасляная кислота), L-карнитин.
- Вальпроаты и Кленбутерол. Исследования показали, что противоэпилептические препараты из группы производных вальпроевой кислоты и агонист бета-адренергических рецепторов Кленбутерол способны увеличивать образование белка выживаемости мотонейронов (SMN) и, соответственно, улучшать клиническое течение заболевания.
- ИПП и прокинетики. Пациентам с гастроэзофагальным рефлюксом, который нередко развивается при нарушении глотания, назначаются ингибиторы протонной помпы (эзомепразол) и средства, стимулирующие моторику желудочно-кишечного тракта (итоприд).
- Муколитики и отхаркивающие средства. У больных со слабостью дыхательной мускулатуры для устранения таких проблем как слабое отхаркивание и скопление в дыхательных путях густой мокроты применяются препараты, разжижающие мокроту (ацетилцисетин) и стимулирующие ее отхаркивание (терпингидрат).
- Гормональные препараты. Людям с возникшими эндокринологическими осложнениями показаны инсулин, сахароснижающие препараты (метформин), тестостерон и антиандрогены (дутастерид).
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Экспериментальное лечение
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни - пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Амиотрофия Верднига-Гоффмана ( Детская спинальная мышечная атрофия , Спинальная мышечная атрофия I типа )
Амиотрофия Верднига-Гоффмана — это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
МКБ-10
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера. Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии
Врожденная форма
СМА I клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
Ранняя детская форма
СМА II дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера
СМА III - наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития, длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Больные спинальной мышечной атрофией I типа находятся под наблюдением детских неврологов и неонатологов. Большое значение имеет возраст манифестации заболевания - для амиотрофии Верднига-Гоффмана характерно развитие с момента рождения до 6 месяцев. В анамнезе часто имеются сведения о позднем и вялом шевелении плода во время беременности.
При осмотре ребенка обращается внимание на выраженную мышечную гипотонию, неспособность самостоятельно сидеть или удерживать голову, типичную «позу лягушки» - плечи приподняты, руки вдоль туловища, ноги выпрямлены и повернуты кнаружи. Отмечаются мышечные подергивания, ослабление или отсутствие сухожильных рефлексов, грубые костные деформации (колоколообразная грудная клетка, Х-образные нижние конечности). Для подтверждения диагноза назначаются следующие дополнительные исследования:
- Лабораторные тесты. В биохимическом анализе крови обнаруживается небольшое повышение концентрации креатинфосфокиназы. У некоторых пациентов данный показатель может находиться в пределах нормальных значений. При анализе газов крови выявляется снижение парциального давления кислорода (paO2) и увеличение углекислого газа (paCO2).
- Спирометрия. Вследствие выраженной мышечной слабости дыхательной мускулатуры при измерении функции внешнего дыхания отмечаются рестриктивные нарушения в виде снижения жизненной емкости легких.
- ЭНМГ. При выполнении игольчатой электронейромиографии удается зафиксировать следующие изменения - резкое уменьшение скорости проведения и амплитуды вызванных потенциалов, спонтанную биоэлектрическую активность в покое (фасцикуляции, фибрилляции).
- Гистология. При патоморфологическом исследовании мышечного биоптата обнаруживается пучковая атрофия мышечных волокон, чередующаяся с неизмененной мышечной тканью, гипертрофированными миофибриллами и соединительнотканными разрастаниями.
- ДНК-анализ. Верифицирующий тест, позволяющий достоверно установить диагноз. Методом полимеразной цепной реакции выявляется генетическая мутация (делеция) SMN1 экзона 7.
Амиотрофию Верднига-Гоффмана следует дифференцировать с другими генетически обусловленными нервно-мышечными заболеваниями, имеющими такое же быстропрогрессирующее течение. К ним относятся врожденные структурные миопатии, ювенильный боковой амиотрофический склероз, синдром Фукуямы.
Лечение амиотрофии Верднига-Гоффмана
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В тяжелых ситуациях (например, при выраженной гипоксемии вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. На сегодняшний день не существует этиотропной терапии амиотрофии Верднига-Гоффмана. Все мероприятия носят симптоматический и паллиативный характер. Применяются следующие методы лечения:
- Обеспечение питания. Так как процесс глотания у многих пациентов затруднен, особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение ребенка - вертикальным. При необходимости устанавливается назогастральный зонд.
- Физиотерапия. Для улучшения метаболизма в мышечных тканях проводятся сеансы электрофореза, электростимуляция модулированным током, грязевые аппликации.
- ЛФК. С целью повышения мышечного тонуса необходимы регулярные физические упражнения - пассивные (выполняются специалистом) с постепенным переходом на активные (выполняются самим пациентом).
- Ортопедическое лечение. Для борьбы с костно-суставными деформациями, а также для их предупреждения используются ортопедические приспособления (корсеты, ортезы, иммобилизирующие шины), фиксирующие различные части тела.
- Респираторная поддержка. Важное место в лечении занимает устранение кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно быть комплексным, проводиться непрерывно и подбираться индивидуально для конкретного пациента. Лекарственные препараты, применяющие для лечения амиотрофии Верднига-Гоффмана следующие:
- Метаболические средства. Для улучшения метаболических процессов в нервных клетках и мышечной ткани назначается коэнзим Q10, L-карнитин, ноотропы. Также для стимуляции регенерации нервной ткани используют высокие дозы витаминов группы В (В1, В6, В12).
- Вальпроаты. Противоэпилептические препараты из группы производных вальпроевой кислоты способны увеличивать образование белка выживания мотонейронов (SMN), что впоследствии может улучшать клиническое течение заболевания.
- ИПП и прокинетики. Ингибиторы протонной помпы (пантопразол) и ЛС, ускоряющие моторику желудочно-кишечного тракта (итоприд), помогают в борьбе с гастроэзофагеальным рефлюксом, который часто возникает у больных АВГ вследствие выраженного нарушения глотания.
- Муколитики и отхаркивающие средства. С целью борьбы с такими дыхательными проблемами как слабое откашливание, скопление в дыхательных путях густой мокроты, применяются препараты разжижающие мокроту (ацетилцистеин) и стимулирующие ее отхаркивание (терпингидрат).
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. У лежачих больных, страдающих постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Новейшие разработки
Ведутся постоянные исследовательские работы по поиску эффективного лекарства для терапии спинальной мышечной атрофии. Наиболее перспективным направлением считается генная терапия. В клинической практике уже используются антисмысловые олигонуклеотиды, исправляющие дефекты матричной РНК в гене SMN2 (Спинраза).
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда — к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Первичная профилактика амиотрофии Верднига-Гоффмана заключается в пренатальной диагностике. При обнаружении в биоптатах ворсин хориона или амниотической жидкости мутации SMN1 показано прерывание беременности. Вторичная профилактика сводится к предупреждению осложнений - аспирационной пневмонии, контрактур суставов, инфекций нижних дыхательных путей.
2. Spinal muscular atrophy: clinical classification and disease heterogeneity/ Russman B.S.// Child Neurology - 2007; 22(8).
3. Regulation of SMN protein stability/ Burnett B.G., Muñoz E., Tandon A. et al.// Mol. Cell. Biol. - 2009. - V. 29. - № 5.
4. A cell system with targeted disruption of the SMN gene: functional conservation of the SMN protein and dependence of Gemin2 on SMN/ Wang J., Dreyfuss G.// J. Biol. Chem. - 2001. - V. 276. - № 13.
Что изменилось в лечении пациентов со СМА в последние годы

Несколько лет назад появилось первое лекарство для пациентов со спинальной мышечной атрофией. Как сегодня получить лечение людям из России и насколько оно эффективно?
В сентябре прошла очередная ежегодная конференция по СМА, организованная благотворительным фондом "Семьи СМА". В этом году повестка конференции существенно отличалась от предыдущей: специалисты рассказывали о новых лекарствах для пациентов со спинальной мышечной атрофией, о том, как они работают и как их можно получить в России. О ключевых посланиях конференции — рассказываем в нашей статье.
Сухие цифры медицинской революции
Впервые болезнь спинально-мышечной атрофии (СМА) описали в конце XIX века, но первая классификация типов заболевания появилась только в 1991 году.
Причину болезни удалось понять 4 годами позже: в 1995-м открыт ген SMN, ответственный за развитие СМА.
До конца 2016 года в мире не существовало лекарства, останавливающего развитие спинально-мышечной атрофии . Никакого.
Сегодня первому препарату 4 года. Это Спинраза (Нусинерсен), — и это уже не единственный вариант терапии. Сегодня в мире для лечения СМА одобрено три лекарственных препарата, в дополнение к уже упомянутой Спинразе — рисдиплам (Эврисди) и генная терапия онасемноген абепарвовек (Золгенсма).
А как у нас?

« В России, как и во всем мире, изменения происходят на глазах, — говорит Ольга Германенко, директор благотворительного фонда «Семьи СМА», член Экспертного совета по редким (орфанным) заболеваниям при Комитете по охране здоровья ГД РФ, член Ассамблеи Европейской Ассоциации «СМА Европа». — Мы очень быстро уходим от позиции «ничего сделать нельзя, положите и не трогайте» к возможностям замедлить течение болезни, а в ряде случаев и получить улучшение состояние здоровья при СМА ».
Наша страна вошла в число государств, где лечат спинально-мышечную атрофию и проводят клинические исследования в этой области. Работа со СМА становится все более «модным направлением».
В августе 2020 были обновлены российские клинические рекомендации по СМА . Что это меняет?
Чем хороши клинические рекомендации-2020
КР — это подробный ответ на вопрос, как выявить и лечить СМА в России.
В варианте-2020 детально описаны основные симптомы СМА каждого типа (кроме СМА4), виды диагностики, жалобы и симптомы, на которые врач (подстрочно: и родитель) обязан обратить внимание; расписаны виды реабилитации для лежачих, сидячих и ходячих пациентов, дан список необходимых технических средств реабилитации (ТСР) и др.
Но главное: в качестве патогенетического лечения рекомендован конкретный препарат — Нусинерсен (Спинраза). Впервые в российской практике. Это значит, что получить лекарство сегодня легче, чем год назад. Аргумент регионального Минздрава «вы назначили слишком дорогой препарат, отзовите назначение!» больше не сработает.
С 2007 года в мире сложился междисциплинарный подход к СМА , отраженный в первых международных рекомендациях по ведению больных СМА, а к 2017 речь идет уже о 10 направлениях терапии.
- Невозможно справляться со СМА только с помощью лекарств, — говорит один из крупнейших экспертов по СМА в Европе Лоран Серве, д.м.с., детский невролог и глава Отдела клинических испытаний и баз данных в Институте миологии при парижском госпитале La Pitie Salpetriere, координатор Института I-Motion, координатор нервно-мышечного центра в Льеже (Бельгия).
СМА — это не только слабость мышц. Сколиоз, периодические эпизоды пневмонии, трудности со сном и приемом пищи часто сопровождают эту болезнь. Нельзя просто принимать Спинразу и на этом успокоиться. Человеку со СМА помимо невролога, педиатра/терапевта и генетика нужен консилиум специалистов. В клинических рекомендациях-2020 он включает:
- специалиста по респираторной поддержке (это пульмонолог и/или анестезиолог-реаниматолог),
- кардиолога,
- травматолога-ортопеда,
- хирурга,
- врача по медицинской реабилитации,
- физиотерапевта,
- диетолога,
- гастроэнтеролога,
- врача паллиативной медицины, при необходимости.
При этом врач-невролог осуществляет основное наблюдение за пациентом.
Привлечь этих и других специалистов — законное право родителей пациента. В том числе с помощью телемедицины, то есть — удаленно.
Спинальная мышечная атрофия (СМА): что это такое?
СМА — редкая генетическая болезнь, требующая участия специалистов паллиативной помощи. Какие степени СМА бывают, чем характеризуется каждая, какое оборудование нужно пациентам - читайте в статье
СМА - одно из самых часто встречающихся генетических заболеваний из разряда редких: оно встречается у одного новорожденного из 6-10 тысяч. Если в семье есть ребенок со СМА, он должен получать паллиативную помощь, поскольку заболевание прогрессирующее и на сегодня - неизлечимое. Однако степени заболевания очень разные: одни пациенты умирают во младенчестве, другие — живут до старости.
Предлагаем вам познакомиться с основной информацией о СМА и той помощи, в которой нуждается семья с этим редким заболеванием. Информацию собрал и упорядочил фонд "Семьи СМА": полную версию "ликбеза", включая истории семей, психологические аспекты, решение медицинских проблем в зависимости от состояния пациента, вы можете найти на сайте специального проекта фонда "Семьи СМА".
Что такое СМА?
СМА (спинальная мышечная атрофия) ― генетическое нервно-мышечное заболевание, которое поражает двигательные нейроны спинного мозга и приводит к нарастающей мышечной слабости. Заболевание носит прогрессирующий характер, слабость начинается с мышц ног и всего тела и с развитием заболевания доходит до мышц, отвечающих за глотание и дыхание. При этом интеллект больных СМА абсолютно сохранен.
В зависимости от тяжести симптомов выделяют 3 основных типа СМА: СМА 1, СМА 2, СМА 3. Чем раньше проявляются первые признаки болезни, тем ярче выражены симптомы, тем они тяжелее и тем быстрее прогрессирует заболевание.
СМА I (БОЛЕЗНЬ ВЕРДНИГА-ГОФФМАНА)
Наиболее тяжелая форма. Возраст проявления болезни: до 6 месяцев
Описание
- Выраженная мышечная гипотония; синдром «вялого ребенка»; не держит голову; не достигает способности сидеть и переворачиваться; обвисшее тело при удерживании подвешенным на животе;
- Ослабленные кашлевой, сосательный и глотательные рефлексы; поперхивание; дыхательные нарушения;
- В анамнезе может быть сниженная внутриутробная активность плода. Может наблюдаться деформация суставов и конечностей из-за внутриутробной гипотонии.
Течение
- Грубая задержка моторного развития;
- Быстрое развитие контрактур и деформаций грудной клетки;
- Прогрессирование бульбарных и дыхательных нарушений, проблемы с глотанием еды и слюны, отхождением мокроты;
- Высокий риск развития аспирационных пневмоний;
- Быстрое нарастание дыхательной недостаточности, особенно при присоединении инфекции.
Прогноз
- Наиболее тяжелая форма: при отсутствии респираторной поддержки большинство детей не доживают до 2 лет;
- Смерть наступает, как правило, из-за нарастания дыхательной недостаточности и развития пневмоний;
- Своевременная респираторная поддержка может увеличить продолжительность жизни ребенка;
- Такие дети нуждаются в паллиативном наблюдении.
«Нет человека без патологических генов». Генетик Наталия Белова рассказывает о редких болезнях и помощи семьям с больными детьми. А также о том, можно ли избежать генетической «поломки» у ребенка и почему жизнь «редких» — пока еще борьба
СМА II (БОЛЕЗНЬ ДУБОВИЦА)
Возраст проявления болезни: 6-18 месяцев.
- Отставание в моторном развитии;
- Способность сидеть без поддержки, иногда ― ползать или стоять, но эти возможности редуцируются по мере взросления;
- Может наблюдаться тремор пальцев;
- Мышечные и скелетные деформации;
- Нарушения дыхания.
- Задержка моторного развития, его остановка и регресс;
- Слабость межреберных мышц, поверхностное диафрагмальное дыхание, ослабление кашлевой функции, со временем развитие дыхательной недостаточности;
- Повышенный риск осложнений после респираторной инфекции;
- Деформации грудной клетки, контрактуры, сколиоз.
- Своевременная помощь и респираторная поддержка увеличивают продолжительность жизни.

Юлия Самойлова. Самый известный в России человек со СМА
СМА III (БОЛЕЗНЬ КУГЕЛЬБЕРГА-ВЕЛАНДЕРА)
Возраст проявления болезни: после 18 месяцев
- Способность ходить самостоятельно (со временем теряется);
- Сложности с комплексными моторными навыками (например, подъем по лестнице, бег);
- По мере прогрессирования болезни могут отмечаться трудности с жеванием и глотанием, а также дыхательные проблемы и проблемы с откашливанием.
- Прогрессирует медленно;
- К подростковому возрасту большинство больных садятся в коляску, но у некоторых способность самостоятельно ходить может сохраниться до взрослого возраста;
- Со временем появляются выраженные контрактуры и сколиоз;
- Риск осложнений после респираторной инфекции.
- При надлежащем уходе имеют обычную продолжительность жизни.
Механизм СМА
СМА вызывается поломкой в гене SMN1. Ген SMN2 частично компенсирует утрату гена SMN1.
- Ген SMN1 поврежден и не соответствует норме
- Важные белки не производятся в достаточном количестве
- Двигательные нейроны функционируют неправильно и отмирают
- Импульсы не распознаются
- Мышцы теряют силу и атрофируются
- Движения ограничены, это затрудняет перемещение, дыхание и глотание
Диагностика и помощь специалистов
При выявлении симптомов, которые могут указывать на болезнь, необходимо комплексное обследование для установления точного диагноза.
Диагностика: необходимо получить консультацию невролога, специалиста по нервно-мышечным заболеваниям, который может установить диагноз на основании симптомов. Для подтверждения диагноза требуется проведение ДНК-диагностики и консультация генетика.
Генетическая диагностика: тест ДНК для выявления делеции гена SMN1 и определения количества копий SMN2.
Дополнительные исследования:
- Биохимия крови: креатинкиназа (КФК) — в норме при СМА I, в норме или незначительно повышена при других типах;
- Электронейромиография — показывает снижение нервных импульсов, помогает дифференцировать СМА от других нервно-мышечных болезней. Сенсорная нервная проводимость обычно в норме.
Медицинская помощь
Врачи-специалисты:
- Невролог ― ставит диагноз, назначает поддерживающее лечение, ведет постоянное наблюдение за ходом болезни.
- Генетик ― ставит диагноз и, в случае необходимости, консультирует семью по вопросам дальнейшего потомства.
- Участковый педиатр (терапевт) ― помогает лечить болезни, которыми болеют все.
- Пульмонолог или реаниматолог ― помогает выявить и скомпенсировать дыхательные нарушения, решать проблемы с откашливанием, дает консультации по респираторной поддержке.
- Ортопед ― оценивает нарушения опорно-двигательного аппарата (контрактуры, деформации), определяет необходимый объем профилактических мер, помогает корректировать эти нарушения.
- Нейрохирург ― занимается исправлением сколиоза. ― подбирает комплекс упражнений и абилитационных процедур и обучает родителей регулярно делать их дома самостоятельно.
- Нутрициолог или диетолог ― помогает подобрать оптимальное питание.
- Гастроэнтеролог ― помогает в случае возникновения проблем с желудком и кишечником.
- Кардиолог ― наблюдает за работой сердечно-сосудистой системы.
- Специалист по паллиативной помощи ― помогает комплексно улучшить качество жизни.
- Другие специалисты привлекаются по мере возникновения специфических проблем.
Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчас Личная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
Важные аспекты:
- Семейно-ориентированный подход ― врач учитывает мнение семьи по всем вопросам, касающимся лечения ребенка, в том числе проведения медицинских вмешательств и их объема, их приемлемости и сроков проведения;
- Ориентация на качество жизни пациента ― прежде чем предложить семье применение каких-либо медицинских технологий, необходимо учесть, как это повлияет на качество жизни, потому что важно жить полноценно, а не существовать;
- Качественная коммуникация и полноценное информирование пациента и членов семьи обо всех аспектах СМА ― как можно более полная информация о болезни, о том, что будет происходить и с чем придется столкнуться, а также как с этим справляться;
- Обучение практическим навыкам ухода и применения медицинского оборудования должно быть неотъемлемой частью медицинской помощи;
- Междисциплинарный и мультипрофессиональный подход ― необходима работа междисциплинарной команды (к примеру, невозможно ограничиться наблюдением невролога и получить весь спектр необходимой помощи для полноценной поддержки).
Помогающие организации
Благотворительный фонд «Семьи СМА» — единственная в России организация, специализирующаяся на помощи семьям, столкнувшимся со спинальной мышечной атрофией. Оказывается благотворительную, информационную, психологическую поддержку семьям, консультирует специалистов по вопросам заболевания и методов работы с пациентами со СМА.
Фонд помощи хосписам «Вера». Благотворительная и консультативная помощь семьям с неизлечимо больными детьми, неизлечимо больным взрослым.
ОДКБ № 1 Отделение паллиативной помощи детям, Екатеринбург, Свердловская область. Оказывается медицинскую, информационную, социальную и психологическую помощь семьям, воспитывающим ребенка-инвалида с паллиативным состоянием.
"Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева" ФГБОУ ВО РНИМУ им.Н.И.Пирогова. Институт находится в Москве. Обращаться за медицинской помощью детям со СМА и другими нервно-мышечными заболеваниями могут жители всей России.
Клиника «Чайка». Консультации врача-пульмонолога Штабницкого Василия Андреевича для детей и взрослых со СМА.
Детский хоспис «Дом с маяком» (Москва, ближнее Подмосковье). Медицинская, психологическая, правовая, социальная, благотворительная помощь семьям с неизлечимо больными детьми и молодыми взрослыми (до 25 лет).
Марфо-Мариинский медицинский центр «Милосердие» (г.Москва). Медицинская, психологическая, правовая помощь, помощь няни, мероприятия, духовная поддержка, благотворительная помощь семьям с неизлечимо больными детьми.
Более подробную информацию вы найдете на сайте благотворительного фонда "Семьи СМА" и их специального проекта о жизни со спинальной мышечной атрофией. Спецпроект предназначен тем, кто столкнулся с диагнозом СМА и хотел бы узнать все самое важное об этой болезни: к каким специалистам и куда обращаться, как ухаживать, за чем следить, о чем помнить, какая терапия существует на сегодняшний день.
Читайте также: