Классификация ВОЗ миелодиспластических синдромов (МДС)
Добавил пользователь Skiper Обновлено: 28.01.2026
Миелодиспластические синдромы (МДС) — группа заболеваний, которые характеризуются нарушениями кроветворения миелоидной линии. В результате этих нарушений возможность выработки зрелых клеток крови частично сохраняется, но наблюдается дефицит тех или иных их видов, а сами клетки при этом изменены и плохо функционируют.
У значительной части больных МДС через некоторый промежуток времени, обычно от нескольких месяцев до нескольких лет, развивается острый миелоидный лейкоз.
МДС в обиходе иногда называют «предлейкемией», ранее применялись также термины «малопроцентный лейкоз», «тлеющий лейкоз» или «дремлющий лейкоз». Это связано с содержанием бластных клеток в костном мозге: если их более 20% (согласно классификации Всемирной организации здравоохранения) или более 30% (согласно франко-американо-британской классификации FAB), то речь уже идет о миелоидном лейкозе, если же их уровень ниже порогового значения, то может быть диагностирован МДС.
Под термином «миелодиспластический синдром» в настоящее время подразумевается целая группа заболеваний, различающихся по частоте встречаемости, клиническим проявлениям, а также по вероятности и ожидаемым срокам трансформации в лейкоз. Специалисты используют две классификации МДС: франко-американо-британскую (FAB) и классификацию Всемирной организации здравоохранения (ВОЗ). Рассмотрим классификацию FAB:
Рефрактерная анемия (РА). Термин «рефрактерная» здесь означает, что анемия не поддается лечению препаратами железа и витаминами. В костном мозге менее 5% миелобластов, аномалии в основном касаются предшественников эритроцитов.
Рефрактерная анемия с кольцевыми сидеробластами (РАКС): миелобластов в костном мозге менее 5%, но не менее 15% предшественников эритроцитов представлены особыми аномальными клетками — так называемыми кольцевыми сидеробластами. Это клетки с кольцеобразными «отложениями» железа, которые не могут обеспечивать эффективный транспорт кислорода.
Рефрактерная анемия с избытком бластов (РАИБ): миелобластов в костном мозге 5-20%. В классификации ВОЗ дополнительно подразделяется на РАИБ-I (5-9% бластов) и РАИБ-II (10-19% бластов).
Рефрактерная анемия с избытком бластов на стадии трансформации (РАИБ-T): миелобластов 21-30% (по классификации ВОЗ это уже острый миелоидный лейкоз).
Хронический миеломоноцитарный лейкоз, ХММЛ (по классификации ВОЗ относится к миелодиспластическим-миелопролиферативным заболеваниям).
Частота встречаемости, факторы риска
Общая частота МДС составляет 3-5 случаев на 100 000 населения. Однако у детей и молодых взрослых это заболевание встречается во много раз реже: более 80% случаев МДС фиксируется после 60 лет, причем несколько чаще у мужчин, чем у женщин.
В большинстве случаев МДС возникает без какой-либо известной причины, но иногда его развитие может быть спровоцировано предшествующей химиотерапией или лучевой терапией по поводу какой-либо опухоли - например, лимфомы Ходжкина или неходжкинской лимфомы. В этих случаях говорят о вторичном МДС.
Частота МДС (как и острого миелоидного лейкоза) повышена у людей с определенными генетическими аномалиями, такими как синдром Дауна, анемия Фанкони, нейрофиброматоз и некоторые другие.
Изучается роль и других факторов в возникновении этого заболевания — например, воздействия некоторых вредных химических веществ. Но у детей и молодых взрослых эти факторы не играют существенной роли.
Признаки и симптомы
Симптомы и степень их выраженности могут различаться в зависимости от разновидности МДС и конкретного случая. Но большинство наблюдаемых проявлений возникает в результате цитопении, то есть дефицита нормальных клеток крови.
Анемия (недостаток эритроцитов, сниженный уровень гемоглобина) характерна для подавляющего большинства случаев МДС; она проявляется бледностью, утомляемостью, плохой переносимостью физических нагрузок; могут также возникнуть одышка, головокружения, боли в груди и т.п.
Примерно у половины больных встречается нейтропения (то есть пониженное содержание зрелых функциональных нейтрофилов) и поэтому снижена сопротивляемость инфекциям. Нередко наблюдается и тромбоцитопения, то есть недостаточное содержание тромбоцитов, что ведет к возникновению кровотечений, синяков, петехий (мелкоточечных подкожных кровоизлияний). Возможны и другие симптомы.
Впрочем, в некоторых случаях пациенты с МДС долгое время не замечают существенного ухудшения самочувствия, и тогда проблемы обнаруживаются только в ходе обычного медосмотра по отклонениям от нормы в клиническом анализе крови.
Диагностика
Как правило, поводом к медицинскому осмотру служат жалобы на симптомы, связанные с анемией, причем эта анемия не поддается обычному лечению препаратами железа и витаминами. В клиническом анализе крови снижено количество эритроцитов, может быть также снижено количество лейкоцитов (нейтрофилов) и/или тромбоцитов. Характерно, что при уменьшении числа эритроцитов их размеры и цветовой показатель крови могут быть увеличены. Полезен также подсчет числа ретикулоцитов — незрелых эритроцитов, так как он дает информацию об интенсивности образования новых красных клеток крови.
Для точной диагностики и определения конкретного варианта МДС необходимо детальное исследование образца костного мозга, взятого в ходе трепанобиопсии: анализируются характер расположения различных клеток («архитектоника» костного мозга), число бластных клеток, содержание кольцевых сидеробластов и других аномальных клеток, степень изменений во всех ростках кроветворения — то есть среди предшественников эритроцитов, гранулоцитов и тромбоцитов, изменения стромы — соединительной ткани костного мозга. Выявленные нарушения могут быть очень разнообразными.
Так как развитие МДС нередко связано с известными хромосомными аномалиями, определенную роль в диагностике и прогнозе играют цитогенетические исследования.
Лечение
Лечение МДС зависит от его конкретной формы. Так, если речь идет об относительно «доброкачественных» разновидностях МДС с небольшим числом бластных клеток, то больные из групп низкого риска могут длительное время сохранять нормальное качество жизни, просто время от времени получая заместительную терапию компонентами крови — эритроцитами и, возможно, тромбоцитами. При перегрузке железом после множественных переливаний необходима соответствующая терапия. Иногда для стимуляции кроветворения используют факторы роста. При инфекционных осложнениях требуется антибактериальная и противогрибковая терапия. В ряде случаев применяют и другие лекарственные средства.
Если же речь идет о формах болезни, связанных с более высоким риском, то вопрос о лечении таких пацентов достаточно сложен. Химиотерапия с использованием обычных цитостатиков (цитарабин и т.п.) малоэффективна и не приводит к долговременной ремиссии. Общепринятых стандартов химиотерапии при МДС практически не существует. Есть и новые лекарства; в частности, обнадеживающие результаты показало применение гипометилирующих препаратов, таких как «Дакоген» (децитабин) и «Вайдаза» (азацитидин). Иногда может применяться иммуносупрессивная терапия и другие методы.
Единственным радикальным методом, позволяющим в случае успеха рассчитывать на полное излечение больных с МДС, является аллогенная трансплантация костного мозга — особенно у молодых пациентов, которые лучше переносят эту процедуру и связанные с ней осложнения. Однако аллогенная трансплантация по поводу МДС, как и по поводу других заболеваний, связана с проблемой поиска совместимого донора и с опасностью жизнеугрожающих осложнений.
При трансформации МДС в острый миелоидный лейкоз проводится химиотерапия этого лейкоза. Однако вторичный лейкоз, развившийся из МДС, обычно плохо поддается терапии. В этой ситуации, как правило, также показано проведение аллогенной трансплантации костного мозга, особенно у молодых больных.
Прогноз
Развитие МДС происходит с разной скоростью в зависимости от конкретной разновидности болезни. Если при некоторых формах МДС пациенты могут, особенно при наличии поддерживающей терапии (переливания компонентов крови и т. д.), жить достаточно долго и полноценно, то при более «активных» и злокачественных разновидностях заболевания средняя продолжительность жизни составляет не более года. Особенно плохой прогноз при вторичном МДС. Пациенты могут погибнуть как от проявлений самого МДС, так и от развившегося на его основе острого миелоидного лейкоза.
При использовании аллогенной трансплантации костного мозга можно добиться нормализации кроветворения и стойкой ремиссии болезни у большинства молодых пациентов. Иными словами, в случае успеха трансплантация приводит к выздоровлению. Однако надо помнить о высокой вероятности жизнеугрожающих осложнений (таких как реакция «трансплантат против хозяина», инфекции и др.) и рецидивов после трансплантации.
Миелодиспластический синдром у взрослых
1. Клинический протокол диагностики и лечения пациентов с заболеванием «миелодиспластический синдром» (далее-МДС) предназначен для оказания медицинской помощи в амбулаторных и стационарных условиях районных, областных и республиканских организаций здравоохранения, имеющих в своем составе гематологические отделения.
2. Возрастная категория: взрослое население.
3. Наименование нозологической формы заболевания (шифр по МКБ-10): миелодиспластический синдром - С92.1;
4. Определение: МДС - группа биологически и клинически гетерогенных клональных заболеваний, характеризующихся неэффективным гемопоэзом и цитопенией в периферической крови вследствие повышения апоптотической активности гемопоэтических предшественников с тенденцией к развитию костно-мозговой недостаточности или острого мие- лобластного лейкоза.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ
5. Классификация МДС, принятая Всемирной организацией здравоохранения (далее-ВОЗ) в 2008 году базируется на цитоморфологических, кариотипических и клинических признаках заболевания.
Классификация миелодиспластических синдромов
- 5 % бластов Палочки Ауэра отсутствуют
- 1 -10 9 /л моноцитов
*- хромосомные аномалии, которые рассматривают как предполагаемое свидетельство наличия МДС при стойкой цитопении неопределенного происхождения и при отсутствии абсолютных морфологических критериев МДС:
несбалансированные аномалии: - 7 или del(7q); - 5 или del(5q); i( 1 7q) или t(17p); - 13 или del(13q); del(l lq); del(12p) или t(12p); del(9q); idic(X)(ql3);
сбалансированные аномалии: t(l 1; 16)(q23;p 13.3); t(3;21) (q26.2;q22.1); t(l;3) (рЗб.З; q21.1); t(2;l 1) (p21;q23); inv(3) (q21q26.2); t(6;9)(p23;q34);
сложный кариотип (3 или более хрмосомных аномалий) с вовлечением вышеупомянутых нарушений.
Диагностика
КРИТЕРИИ ДИАГНОЗА МДС
6. Предварительные критерии (А).
6.1. Стабильная цитопения по > 1 из следующих клеточных линий:
эритроидной (уровень гемоглобина < 110 г/л);
нейтрофильной (количество нейтрофилов < 1,5х10 9 /л);
мегакариоцитарной (количество тромбоцитов < 100 х10 9 /л).
6.2. Исключение других гематологических и негематологических заболеваний - причин цитопении/дисплазии.
7. МДС-ассоциированные критерии (В).
7.1. Дисплазия в > 10% клеток эритроидной, нейтрофильной или мегакариоцитарной клеточных линиях костного мозга, либо наличие > 15% кольцевых сидеробластов.
7.2. От 5 до 19% бластных клеток в костном мозге.
7.3. Типичные хромосомные абберации по результатам стандартного цитологического исследования или флюоресцентной гибридизации in situ (+8, -7, 5q-, 20q- и др. согласно классификации)
8. Вспомогательные критерии (С) (для пациентов, имеющих критерии А, но не имеющих критерии В).
8.1. Аномальный иммунный фенотип эритроидных или миелоидных клеток костного мозга, указывающий на их клональное происхождение (по результатам проточной цитометрии).
8.2. Молекулярно-генетические признаки наличия клональной клеточной популяции в костном мозге (по результатам HUMARA исследования или биологического микрочипирования).
8.3. Значительное и стабильное снижение колониеобразующей активности костного мозга и/или периферической крови.
Диагноз устанавливается при наличии 2 предварительных критериев (А) и не менее чем одного МДС-ассоциированных критериев (В). Вспомогательные
критерии (С) используются при отсутствии критериев В и наличии у пациента признаков клональной миелоидной пролиферации. Критерии группы С не входят в обязательный стандарт диагностики МДС.
Диагноз «идиопатическая цитопения неопределенного значения» применяется для обозначения случаев цитопении по одной и более клеточным линиям в течение > 6 месяцев при отсутствии критериев МДС и других причин цитопении. Такие пациенты должны наблюдаться и обследоваться гематологом с интервалом 1-6 месяцев.
Диагностические критерии разработаны ICWG (International Consensus Working Group), 2007 г.
Алгоритм диагностики МДС включает в себя клинические и лабораторные исследования, мультидисциплинарный подход с привлечением смежных специалистов и последовательно проводится на базе учреждений здравоохранения различного уровня с соблюдением преемственности на всех этапах. Это обусловлено полиэтиологичностью и гетерогенностью проявлений при данной патологии, стремлением к рационализации использования специального диагностического оборудования, минимизации диагностических ошибок.
Этапы диагностики МДС:
| Этап | Задачи | Уровень медицинского учреждения и специалисты | Содержание |
| Первичного скрининга | Выявление пациентов с цитопе- ническими синдромами и клиническими проявлениями МДС; обоснование необходимости и направле-ние пациентов, нуждающихся в дополнительном обследовании, на следующий этап | Районный, городской (врачи любой специальности амбулаторно - поликлинических и стационарных учреждений здравоохранения) | Анамнез (химио- или лучевая терапия в прошлом, случаи МДС/ОМЛ в семье, рецидивирующие инфекционные заболевания или геморрагический синдром) Физикальное обследование (бледность, инфекционновоспалительные процессы, геморрагический синдром, спле- номегалия) Общий анализ периферической крови, с подсчетом количества тромбоцитов, лейкоцитарной формулы. Биохимический анализ крови (общий белок, альбумины, глобулины, мочевина, креатинин, билирубин, щелочная фосфатаза, ACT, АЛТ, ЛДГ, сывороточное железо) |
| Углубленного кли- нико - лабораторного и инструментального обследования | Углубленное клиниколабораторное и инструментальное обследование и выявление МДС - ассоциированных критериев; обосно-вание необходимости и направление па-циентов на следующий этап; диспансеризация пациентов с ранее верифицированным МДС | Областной, городской (гематоло гические отделения, кабинеты) | Общий анализ периферической крови, с подсчетом количества тромбоцитов, ретикулоцитов, лейкоцитарной формулы, морфологической оценкой. Биохимический анализ крови (сывороточный ферритин) Исключение реактивной дисплазии (мегалобластная анемия в результате дефицита витамина В12 и фолиевой кислоты, инфицирование ВИЧ, алкоголизм, недавнее цитотоксическое лечение, солидные злокачественные новообразования): маркеры вирусных гепатитов В и С, сифилиса, вируса иммунодефицита человека, ФГДС, колоноскопия, ультразвуковое исследование органов брюшной полости и малого таза, лимфатических узлов, щитовидной железы, рентгенография органов грудной полости. Аспирационная биопсия костного мозга: морфологическое исследование, иммунный фенотип, цитохимическое исследование эритрокариоцитов |
| Дифференциальной диагностики и подбора терапии | Клинико - лабораторное и инструментальное обследование пациентов с целью дифференциальной диагностики, верификации диагноза; подбор и коррекция индиви-дуальной про-граммы терапии; диспансеризация пациентов с ранее верифицированным МДС; создание базы и анализ данных пациентов с МДС для изучения проблемы МДС и усовершенствования оказания медицинской помощи | Областной, республи канский (гематоло гические отделения) | Исключение реактивной дисплазии: маркеры вируса Эпштейна - Барр, цитомегаловируса, определение в сыворотке крови уровня витамина В]2 и фолиевой кислоты. Определение в сыворотке крови уровня эритропоэтина Аспирационная биопсия костного мозга: морфологическое, цитогене- тическое исследования, иммунный фенотип, цитохимическое исследование эритрокариоцитов костного мозга Билатеральная трепанобиопсия передних или задних остей подвздошных костей Молекулярно-биологический анализ Клоногенный тест |
Лечение
КЛИНИЧЕСКИЕ ВАРИАНТЫ МДС
10. Определение клинического варианта МДС имеет значение для выбора тактики лечения.
10.1. 5ц-синдром: болеют преимущественно женщины, характерны вялотекущий характер заболевания, низкая вероятность трансформации в ОМЛ (10%), тяжелая макроцитарная анемия, нормальный или умерено сниженный уровень лейкоцитов и тромбоцитов, дисплазия мегакариоци- тарного ростка, отсутствие значительно повышения уровня бластных клеток в костном мозге; хороший ответ на леналидомид*.
10.2. Вторичный МДС: частота вторичного МДС нарастает в связи с успехами химиотерапии опухолей и воздействием загрязнения окружающей среды; для большинства пациентов характерны множественные хромосомные аберрации; прогноз хуже, чем при первичном МДС.
10.3. Гипопластический МДС:
до 15% случаев МДС характеризуются низкой клеточностью костного мозга при гистологическом исследовании (доля кроветворной ткани в препарате менее 30% у пациентов моложе 60 лет или менее 20% у пациентов 60 лет и старше);
дисплазия мегакариоцитов и клеток миелоидного ряда может отсутствовать;
возможны трудности в дифференциации от апластической анемии, для которой характерна более выраженная панцитопения, отсутствие типичных для МДС хромосомных аббераций и снижение содержания CD34+ клеток в костном мозге.
10.4. МДС с миелофиброзом: до 50% случаев всех вариантов МДС характеризуется фиброзом костного мозга (до 15% имеют выраженный фиброз); фиброз более характерен для вторичного МДС; характерны ги- перклеточность костного мозга, диффузный ретикулиновый фиброз его стромы и дисплазия не менее чем в 2 клеточных линиях; в периферической крови панцитопения, признаки клеточной дисплазии и лейкоэрит- робластоза; органомегалия нехарактерна; заболевание быстро прогрессирует; необходимо дифференцировать от острого мегакариобластного лейкоза, острого миелофиброза (острого панмиелоза с фиброзом), хронических миелопролиферативных заболеваний, метастатического рака, лимфом и волосатоклеточного лейкоза.
ЛЕЧЕНИЕ
11. Выбор терапии основан на диагнозе и группе риска по международной прогностической бальной системе (IPSS). В соответствии с международными рекомендациями для выбора терапевтической тактики пациентов с МДС подразделяют на 2 большие группы риска:
группу относительно низкого риска, включая в нее пациентов с низким и промежуточным 1 риском по системе IPSS;
группу высокого риска, включая в нее пациентов с промежуточным 2 и высоким риском по системе IPSS.
У пациентов из группы относительно низкого риска возможно применение только поддерживающей терапии либо терапии малой интенсивности. Интенсивная терапия показана пациентам группы высокого риска с учетом возраста, анамнеза заболевания, клинических проявлений, общего состояния и наличия признаков прогрессирования заболевания.
11.1. Поддерживающее лечение.
Поддерживающее лечение назначают с целью уменьшения проявлений заболевания и поддержания качества жизни. У пациентов из группы относительно низкого риска это может быть основным видом терапии.
11.1.1. Трансфузии донорских эритроцитов. Основным клиническим показанием для трансфузии донорских эритроцитов является не столько уровень гемоглобина, сколько степень адаптированности пациента к анемии.
11.1.2. Применение хелаторов железа.
Показаниями к применению хелаторов железа является переливание более 20-25 доз эритроцитной массы, уровень сывороточного ферритина более 2500 мкг/л, наличие дисфункции сердца (аритмия, сердечная недостаточность) и поражения печени.
Дефероксамин применяют в дозе 30-40 мг/кг в виде 12 часовых подкожных инфузий 5-7 раз в неделю (ночью). Дозу лекарственного средства снижают до 25 мг/кг при уровне ферритина < 2000 мкг/л. Необходимы контроль функции почек, аудиометрия и офтальмологический контроль до начала терапии и ежегодно на фоне ее проведения.
11.1.3. Трансфузии донорских тромбоцитов в стандартных дозировках показаны пациентам с глубокой тромбоцитопенией и петехиально - пятнистой кровоточивостью.
11.1.4. Эмпирическая антибактериальная и противогрибковая терапия лекарственными средствами широкого спектра действия показана пациентам с фебрильной нейтропенией. Профилактический прием антибактериальных и противогрибковых лекарственных средств показан лишь пациентам с рецидивирующими инфекционными осложнениями на фоне нейтропении.
11.2. Терапия малой интенсивности.
11.2.1. Эритропоэтин применяют в качестве терапии первой линии у пациентов группы относительно низкого риска с РА и РАИБ, частота трансфузий донорских эритроцитов у которых менее 2 доз в месяц и базальный уровень эритропоэтина в сыворотке крови менее 200 МЕ/л, в дозе 10 000 Ед подкожно в сутки ежедневно (40-60 000 ЕД 1-3 раза в неделю) в течение 6 недель.
11.2.2. Филграстим (далее - Е-КСФ) назначают пациентам резистентным к монотерапии эритропоэтином в дозе 1-2 мкг/кг подкожно в сутки ежедневно или 1-3 раза в неделю (в сочетании с эритропоэтином). При отсутствии ответа на терапию в течение 2-3 месяцев её прекращают. При наличии ответа постепенно снижают дозу эритропоэтина и Е-КСФ до минимально эффективной.
Возможно монотерапия Е-КСФ у пациентов с нейтропенией и рецидивирующими или резистентными к антибиотикотерапии инфекциями. Профилактическое применение препарата не целесообразно.
11.2.3. Эпигенетическую терапию применяют у пациентов группы высокого риска, у которых невозможно применение интенсивной терапии:
децитабин 20 мг/м в сутки внутривенно 5 дней ежемесячно 4-6 курсов.
11.2.4. Иммуносупрессивную терапию применяют преимущественно у пациентов с гипопластическим вариантом МДС.
Антитимоцитарный глобулин (далее-АТГ) 40 мг/кг в сутки внутривенно 4 дня.
Циклоспорин А в дозе 1-5 мг/кг/день в 2 приема не менее 6 месяцев. Дозу корригируют в соответствии с концентрацией препарата в сыворотке крови (не выше 400 мкг/мл), уровнем артериального давления, функциональным состоянием печени и почек.
11.2.5. Пациентам с РАИБ-1 и РАИБ-2, гипопластическим вариантом МДС показан мелфалан в дозе 2 мг/сутки перорально до получения клинико-гематологического эффекта.
11.3. Интенсивная терапия.
11.3.1. Пациентам группы высокого риска в возрасте менее 60 лет показано применение терапии индукции ремиссии острого миелобластно- го лейкоза.
11.3.2. Высокодозная химиотерапия с трансплантацией аллогенных гемопоэтических стволовых клеток показана всем пациентам с МДС в возрасте менее 60 лет при наличии HLA-идентичного родственного донора.
11.3.3. Критерии клинико-гематологического ответа при лечении первич
Миелодиспластический синдром
Миелодиспластический синдром - группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение - переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Общие сведения
Миелодиспластический синдром - группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома - курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие - при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким - около 8 месяцев. Вероятность отторжения костного мозга после трансплантации - около 10%.
Миелодиспластический синдром: классификация, развитие, лечение, рекомендации, прогноз
Миелодиспластический синдром (МДС) - это не одна какая-то болезнь, это целая группа различных патологических состояний костного мозга (КМ), отнесенных к гематологии, но пока не причисленных к лейкозам, хотя болезнь оставляет высокий риск перехода в более тяжелую форму (лейкоз).
Суть МДС заключается в нарушении костномозгового кроветворения на миелоидной линии в отношении какого-то одного клона клеток или затрагивающего несколько популяций. В любом случае для миелодиспластического синдрома характерным признаком будет изменение качественного и количественного состава периферической крови.
Коротко о гемопоэзе
Кроветворение (гемопоэз) - процесс, проходящий много стадий, на каждой из которых клетки крови приобретают новые качества (дифференцируются). Конечным результатом этого процесса является выход в периферическую кровь зрелых (или созревающих, но уже имеющих определенные «навыки»), полноценных, способных осуществлять свои функциональные задачи, форменных элементов крови:
- Красных кровяных телец - эритроцитов;
- Белых клеток - лейкоцитов;
- Кровяных пластинок (бляшек Биццоцеро) - тромбоцитов.
Кроветворение начинается от стволовой клетки, способной, дифференцироваться и давать жизнь всем линиям (росткам) гемопоэза. Миелоидный и лимфоидный ростки пошли от специализированных, обладающих высокой пролиферативной активностью, способных к дифференцировке плюрипотентных клеток.
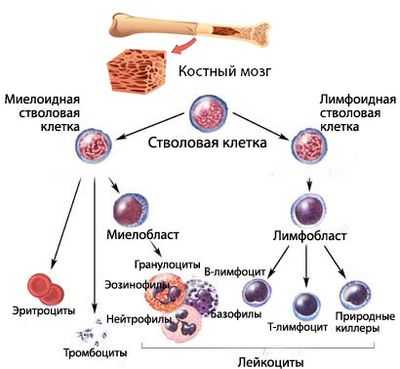
Сбой кроветворения в миелоидном направлении приводит к тому, что сам аномальный клон в некоторой степени теряет возможность продолжать линию (воспроизводить потомство, поэтому количество клеток того ростка, на котором возникла проблема, падает). Естественно, нарушается и созревание полноценных клеток. В результате этого, уменьшается численность одной или нескольких популяций форменных элементов, а также, ввиду ухудшения качества клеток, не в лучшую сторону меняются их функциональные возможности.
Вытекающие из подобных событий последствия - синдром, имеющий различные варианты клинических проявлений, то есть, представляющий собой группу гетерогенных патологических состояний, которая и названа миелодиспластическим синдромом.
Позиция МДС в Международной классификации болезней
Международная классификация болезней десятого пересмотра (МКБ-10), принятая Всемирной организацией здравоохранения (ВОЗ) в Швейцарии Женева, 1989), вступила в силу на территории Российской Федерации в 1997 году. Между тем, в отношении многих патологических состояний в 2010 году были внесены изменения. Нововведения коснулись и гематологической патологии, в том числе, миелодиспластического синдрома. По МКБ-10 в блок диагнозов D37-D48 МДС входит под своим кодом - D46, который имеет 7 или 9 вариантов определений заболеваний или диагнозов (в России, наряду с классификацией ВОЗ, могут использоваться и другие классификации, например, FAB, где вообще только 5 вариантов, поэтому в разных справочниках кодирование также может иметь отличия):
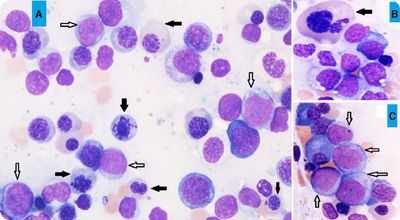
пример МДС с избытком бластов
Примечание: так часто встречающееся определение «рефрактерная» в данном случае объясняет безуспешность лечения железосодержащими и витаминными лекарственными средствами. Рефрактерная анемия устойчива к подобным мерам воздействия, не реагирует на них и нуждается в других терапевтических мероприятиях.
Общая характеристика синдрома
Аномалия генетического материала на уровне полипотентной кроветворной стволовой клетки, мутация ее, а также клеток предшественниц кроветворения, наличие генетически неполноценных клонов приводят к тому, что в клеточном звене системы иммунитета происходят существенные изменения, глубина которых, однако, зависит от того, по каким линиям (одной или нескольким?) пошли нарушения в кроветворении. В зависимости от этого можно ожидать в крови:
- Моноцитопению (уменьшение клеток одного вида);
- Бицитопению (нарушения идут в двух ростках);
- Панцитопению (сбой пошел в трех направлениях, поэтому резко снижено количество белых и красных клеток крови, а также тромбоцитов).
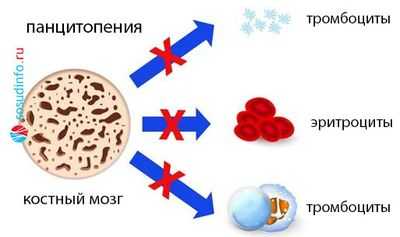
В КМ - аналогично: нормоклеточность, гиперклеточность либо гипоклеточность (миелограмма покажет, какой росток пострадал).
Клинические проявления описываемого синдрома также соответствуют причине, спрятанной на уровне кроветворения:
- Анемия;
- Геморрагический синдром (при снижении численности и нарушении функции тромбоцитов);
- Сочетание анемического и геморрагического синдромов;
- Инфекционный синдром (реже);
- Увеличение селезенки, лимфаденопатия, постоянное повышение температуры тела (эти симптомы присутствуют не так и часто, поэтому относятся к факультативным признакам).
Между тем, опираясь на данные многочисленный исследований МДС (изменение численности и морфологических характеристик клеток крови и костного мозга), гематологи пришли к выводу, что рано или поздно конечным итогом миелодиспластического синдрома станет острый или хронический миелоидный лейкоз (ОМЛ либо ХМЛ), а все эти анемии (рефрактерные) являются лишь промежуточным (временным) состоянием болезни. В связи с этим МДС нередко называют «предлейкозом», «предлейкемией», «тлеющим» или «дремлющим» лейкозом. Все зависит от количества миелобластов - клеток-родоначальниц гранулоцитарного ряда.
Если рефрактерная анемия протекает с избытком бластов (>20% по данным ВОЗ или >30% согласно классификации FAB), то гематологи склоняются к диагнозу - миелоидный лейкоз. В ситуациях, когда численность бластных клеток вплотную не подходит к этому порогу, диагноз пациента остается прежним - миелодиспластический синдром.
Патологическое состояние главного кроветворного органа может сформироваться у человека в любом возрастном периоде (от грудного - до глубокой старости). У детей болезнь чаще всего дебютирует между 3 и 5 годами, хотя, в целом, в детском возрасте риск заболеть совсем низкий. Среди взрослых самыми уязвимыми становятся пожилые люди (60 лет и старше). Например, такой распространенной и рискующей перейти в острый лейкоз форме, как РЦМД, наиболее подвержены люди в возрасте от 70 до 80 лет. Общая частота встречаемости миелодиспластического синдрома колеблется в пределах 3-5 случаев на 100 тысяч населения (не так и редко), причем, мужчины страдают данной патологией несколько чаще, нежели женщины.
Причина первичных форм заболевания остается невыясненной. Основными вероятными «виновниками» вторичного МДС считаются:
- Воздействие ионизирующего излучения;
- Влияние антропогенных неблагоприятных факторов окружающей среды (химических соединений, созданных человеком);
- Последствия химио- и радиотерапии (после лечения опухолевых процессов);
- Инфекционные агенты (бактерии, вирусы).
Следует отметить, что до сих пор МДС, передаваемого по наследству или возникающего в кругу близких родственников, отмечено не было, однако из наблюдений определена группа пациентов, имеющих повышенный риск формирования синдрома. Это дети и взрослые люди, страдающие болезнью Дауна, анемией Фанкони, синдромами Луи-Бар и Блума.
Лечатся все по-разному
Следует сразу настроить пациента, что лечение МДС не будет одинаковым для всех его разновидностей. Набор терапевтических мероприятий рассматривается в индивидуальном порядке, исходя из формы болезни и категории риска, которой принадлежит пациент (согласно клинической классификации Международной Прогностической Системы - IPSS для МДС: низкий, промежуточный 1 и 2, высокий). Словом, существуют определенные каноны, которых доктор придерживается, прежде чем приступить к непосредственному лечению. К примеру:
- Люди, не перешагнувшие 60-летний рубеж, имеющие минимальные признаки болезни, но отнесенные к категории промежуточного или высокого риска с ожидаемой выживаемостью 0,3 - 1,8 года, подвергаются высокоинтенсивной терапии;
- Пациенты, принадлежащие к группе промежуточного и низкого риска с ожидаемой выживаемостью 5-12 лет, проходят лечение низкой интенсивности;
- Молодые люди и больные среднего возраста (до 60 лет) с относительно неплохими показателями (ожидаемая выживаемость от полугода до 5 лет) первоначально лечатся по схемам низкой интенсивности, хотя в любой момент им грозит оказаться в группе, получающей более жесткое лечение (высокие дозы химиотерапии, пересадка КМ).
Таким образом, схемы лечения миелодиспластического синдрома довольно сложны и знает их только врач, получивший в свое время определенную специализацию (гематолог). Он в своей лечебной тактике опирается на рекомендации, разработанные Британским комитетом по стандартизации в гематологии (редакция 2009 года). Читателю же, на наш взгляд, достаточно познакомиться с основными методами проведения терапевтических мероприятий, особо не вникая в тонкости, не ставя диагноз и не причисляя себя или своих близких к той или иной группе риска. И еще, наверное, не помешает знать, что:
- Лечение высокой интенсивности - это, во-первых, обязательное пребывание в специализированном стационаре, во-вторых, назначение высоких доз химиотерапии и, возможно, подготовка к пересадке стволовых клеток и сама пересадка;
- Низкоинтенсивная терапия подразумевает пребывание в больнице (или даже в условиях дневного стационара) время от времени для получения заместительной терапии, низких доз химиопрепаратов, симптоматического лечения.
К сожалению, способа избавиться от такого тяжелого недуга, как МДС, раз и навсегда, пока не придумали. Разве что пересадка главного кроветворного органа (костного мозга) могла бы решить проблему, однако она тоже сопряжена с определенными трудностями (иммунологическое типирование, поиск совместимого донора, высокая стоимость операции, если искать донора по всему миру). Правда, в последние годы, как на территории Российской Федерации и ближайшей соседки - Беларуси, так и на территории других государств бывшего СССР, создаются новые лаборатории тканевого типирования, объединяющие свои реестры в единый банк, чтобы иметь возможность помочь друг другу. На них и возлагаются будущие надежды.

Если врач считает, что патологический процесс идет как бы доброкачественно (если можно так выразиться), с небольшим количеством бластов, то больные группы низкого риска, периодически получающие заместительное и поддерживающее лечение (эритроцитарную массу, тромбовзвесь), могут довольно продолжительное время работать и вести почти привычный образ жизни. В основном, лечение таких больных выглядит следующим образом:
- Больной направляется в стационар, чтобы не допустить значительного снижения гемоглобина и развития тяжелого анемического синдрома, поэтому борьбу с ним (анемическим синдромом) считают первостепенной задачей (переливание эритроцитарной массы, заготовленной от доноров);
- Не упускается из виду и такое проявление МДС, как геморрагический синдром, возникающий на почве снижения числа и функциональной неполноценности тромбоцитов. В принципе, симптоматическая терапия, которая позволяет удерживать количество форменных элементов на нужном уровне (гемотрансфузии - эрмасса, тромбовзвесь и т. д.), в общем-то, всегда присутствует в схеме лечения больных, имеющих относительно благоприятную форму болезни;
- Получая от случая к случаю донорские эритроциты, организм больного начинает перегружаться железом, что ликвидируется применением медикаментозных средств, образующих комплексы с этим химическим элементом (эксиджад, десферол);
- Иной раз больные нуждаются в назначении низких доз «химии» (цитарабин, децитабин), а также иммунодепрессивных средства для предотвращения иммунной агрессии против костного мозга (леналидомид), с добавлением к ним ATG (антимоноцитарный глобулин) и циклоспорина;
- Присоединение инфекционного агента требует лечения антибиотиками и противогрибковыми препаратами.
Гораздо сложнее лечить формы миелодиспластического синдрома с избытком бластов, входящие в категорию высокого риска, когда химиотерапевтические препараты почти не приносят желаемого результата и не «отправляют» больного в долгосрочную ремиссию. Однако это не значит, что от них отказываются вовсе, ведь новые, недавно разработанные лекарства, дают некоторую надежду в отношении МДС и даже применяются для лечения ОМЛ (острого миелобластного лейкоза). Однако при таких обстоятельствах существуют рекомендации разработчиков - применять подобные средства для лечения больных, не достигших 60-летнего возраста и имеющих неплохой иммунологический статус, в противном случае - есть риск развития серьезных осложнений, способных преждевременно прервать жизнь.
Пересадка стволовых клеток (возможна тоже только до 60 лет) на сегодняшний день - единственный способ избавить человека от страданий на долгие-долгие годы. К сожалению, трансплантация КМ - операция хоть и несложная в техническом плане, но трудновыполнимая в плане подбора по лейкоцитарной системе HLA совместимого с реципиентом (больным) донора (идентичными, то есть, имеющими абсолютно одинаковый набор генов являются только однояйцевые близнецы - это идеальные доноры друг другу).
Частные симптомы и диагностика
Клинические проявления и степень их выраженности по причине многообразия форм МДС позволяют себе широкие вариации. Случайной находкой синдром выступает редко (это бывает, если человек неплохо себя чувствует, а анализы назначаются в силу других обстоятельств). В основном же, больные направляются в поликлинику с определенными жалобами (постоянное ощущение усталости, одышка, физическая слабость, головокружения, частые подъемы температуры тела), где после тестирования крови становятся очевидными и другие признаки миелодиспластического синдрома:
- Цитопения (снижение количества полноценных форменных элементов крови);
- Анемия (низкий гемоглобин, мало эритроцитов), которая и определяет симптомы, заставившие пойти к врачу;
- Нейтропения (недостаточное содержание в крови нейтрофильных лейкоцитов, обладающих способностью поглощать бактериальные клетки в очаге воспаления - она становится причиной частых инфекций и лихорадки);
- Тромбоцитопения (уменьшение численности тромбоцитов, что обуславливает появление геморрагического синдрома - кровотечений, мелкоточечных подкожных кровоизлияний, синяков).
Между тем, отдельные пациенты относительно долго могут жить и не подозревать, что здоровье «пошатнулось». И тогда МДС становится случайной находкой уже на стадии проведения общего анализа крови.
Чаще всего поводом все же обратиться в поликлинику служат жалобы больного, которые в наибольшей степени связаны с анемией. Пробовать повысить уровень красного пигмента крови (Hb) и содержание красных кровяных телец (Er) препаратами железа и витаминами бесполезно, лечение успехов не приносит, ведь анемия при МДС - рефрактерная. При подозрении на МДС, которое возникает в ходе проведения общего анализа крови (ОАК), добавляются другие исследования:
- Подсчет молодых форм красного ростка, которым уже «позволено» присутствовать в циркулирующей крови - ретикулоцитов, они «подскажут», насколько быстро идет процесс воспроизводства новых зрелых кровяных телец;
- Цитологическое исследование аспирата КМ (у пожилых пациентов данный тест не принадлежит к обязательным анализам);
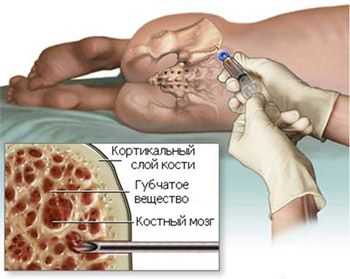
трепанбиопсия костного мозга
Трепанобиопсия (процедура обязательна для всех больных) - после изучения морфологических особенностей гистологический анализ развеет сомнения или подтвердит подозрения;
Безусловно, диагностика миелодиспластического синдрома, начинается с жалоб больного и ОАК, но в дальнейшем опирается на более сложные лабораторные исследования. Здесь врачу есть над чем подумать, чтобы правильно оценить нарушения кроветворения, ведь изменения клеточного состава и морфологических особенностей клеток крови и костного мозга могут быть весьма многочисленны и многообразны. Впрочем, как и сама болезнь…
костный мозг при МДС
Прогноз и рекомендации
Прогноз в отношении продолжительности жизни при миелодиспластическом синдроме не очень оптимистичный, хотя многое зависит от разновидности болезни, степени риска и возрастной категории больного. В целом, пациенты, строго выполняющие рекомендации лечащего врача и получающие периодически поддерживающее лечение, могут рассчитывать прожить пять, а то и десять лет. Однако активное течение злокачественной формы болезни оставляет мало шансов - если не был найден донор и не пересажена стволовая клетка, жизнь может прерваться на 1-2 году от начала патологического процесса. Причиной смерти в большинстве случаев становится острый миелоидный лейкоз, который развился на почве вторичного МДС.
В заключение хочется дать совет людям, столкнувшимся с подобной проблемой и желающим продлить свою жизнь или жизнь близким: никогда не слушать рекомендации того, кто почерпнул сведения о болезнях из сомнительных источников (подобная информация вовсю «гуляет» на просторах Интернета) и возомнил себя доктором. Ни народными средствами, ни специальными физическими упражнениями миелодиспластический синдром не лечится. Нужно следовать рекомендациям врача и тогда, возможно, лечение будет успешным.
Миелодиспластические синдромы (МДС)
Миелодиспластические синдромы (МДС) - это группа злокачественных заболеваний, которые происходят из тканей костного мозга и влияют на рост клеток крови, что приводит к нарушению нормального функционирования организма. Примерно в каждом третьем случае МДС переходит в острый миелоидный лейкоз (ОМЛ) - заболевание, при котором клетки крови начинают бесконтрольно размножаться.
Первоначально миелодиспластические синдромы рассматривались как предлейкозные состояния и были классифицированы как отдельные нарушения только в 1976 году. В общей совокупности населения МДС встречается относительно редко, однако среди пожилых людей старше 60 лет заболеваемость составляет 20-50 случаев на каждые 100000 человек.
Что такое миелодиспластические синдромы?
При миелодиспластических синдромах стволовые клетки крови не развиваются должным образом и не превращаются в здоровые тромбоциты, эритроциты или лейкоциты. Поскольку данные клетки остаются незрелыми, они не могут нормально функционировать и погибают либо в костном мозге, либо вскоре после попадания в кровоток. Вследствие этого в организме пациента развивается нехватка здоровых клеток крови.
Каковы факторы риска развития миелодиспластических синдромов?
Существует несколько известных факторов риска развития миелодиспластических синдромов:
- Пожилой возраст: в большинстве случаев МДС диагностируется у пожилых людей в возрасте от 70 до 80 лет.
- Пол: МДС чаще встречается у мужчин, чем у женщин. Причина этого явления неизвестна.
- Предшествующая химиотерапия онкологического заболевания:лечение рака некоторыми химиотерапевтическими препаратами повышает риск развития МДС.
- Генетические синдромы: пациенты с некоторыми наследственными синдромами имеют более высокий риск развития МДС. С развитием МДС связаны следующие генетические заболевания: анемия Фанкони, синдром Швахмана-Даймонда, анемия Даймонда-Блекфена, семейная тромбоцитопатия с предрасположенностью к развитию миелоидных злокачественных опухолей, тяжелая врожденная нейтропения и врожденный дискератоз.
- Наличие МДС в семейном анамнезе: определенная генетическая мутация, присутствующая в семейном анамнезе, может повышать риск возникновения МДС.
- Курение: канцерогенные вещества, содержащиеся в табачном дыме, всасываются в кровь при ее прохождении через легкие и распространяются по всему организму.
- Воздействие факторов окружающей среды: длительный контакт с химическими веществами, которые используются в резиновой и нефтяной промышленности (бензол и др.), может повышать риск развития МДС. Кроме того, воздействие высоких доз радиации (как, например, при аварии на ядерном реакторе) также повышает риск развития заболевания.
Каковы симптомы миелодиспластических синдромов?
Миелодиспластические синдромы приводят к снижению уровней показателей крови, поэтому ранние симптомы болезни связаны с дефицитом одного или нескольких типов клеток крови.
Симптомы миелодиспластических синдромов включают:
- Анемия (пониженное содержание эритроцитов) может вызвать следующие симптомы:
- Усталость;
- Головокружение;
- Слабость;
- Бледность кожного покрова;
- Одышка;
- Головные боли.
- Лейкопения (пониженное содержание лейкоцитов) может вызвать следующие симптомы:
- Частые или тяжелые инфекционные заболевания.
- Тромбоцитопения (пониженное содержание тромбоцитов) может вызвать следующие симптомы:
- Кровоточивость и/или появление синяков при незначительном воздействии;
- Частые или сильные носовые кровотечения;
- Кровоточивость десен.
Дополнительные симптомы:
- Повышенная температура тела;
- Боли в костях;
- Потеря веса;
- Отсутствие аппетита.
Типы миелодиспластических синдромов
Существует несколько типов МДС, различающихся по степени поражения клеток крови и костного мозга. Согласно системе Всемирной организации здравоохранения (ВОЗ), классификация МДС осуществляется на основе внешнего вида клеток под микроскопом и количества аномальных клеток.
Система классификации ВОЗ выделяет шесть основных типов миелодиспластических синдромов:
- МДС с мультилинейной дисплазией: наблюдается поражение двух или более типов клеток крови.
- МДС с однолинейной дисплазией: поражены преимущественно эритроциты.
- МДС с кольцевыми сидеробластами: эритроциты не могут перерабатывать железо, необходимое для выработки гемоглобина. Вместо этого молекулы железа образуют кольцо вокруг ядер развивающихся эритроцитов.
- МДС с избытком бластов: наблюдается поражение одного или более типов клеток крови с избыточным содержанием бластных клеток в крови и в костном мозге.
- МДС с делецией 5q: наблюдается поражение эритроцитов, вызывающее анемию. В костном мозге и в крови присутствует небольшое количество бластных клеток.
- МДС неклассифицируемый.
МДС также классифицируется на основе клинической картины, относящейся к первопричине болезни. Если причина не установлена, то МДС называется первичным (наиболее распространенный тип). Когда причина заболевания известна - это вторичный МДС. Диагностика типа МДС играет важную роль в выборе оптимального курса лечения.
Что такое МДС?
Каковы факторы риска развития МДС?
Существует несколько известных факторов риска развития миелодиспластических синдромов.
- Пожилой возраст: в большинстве случаев МДС диагностируется у пожилых людей в возрасте от 70 до 80 лет.
- Пол: МДС чаще встречается у мужчин, чем у женщин. Причина этого явления неизвестна.
- Предшествующая химиотерапия онкологического заболевания: лечение рака некоторыми химиотерапевтическими препаратами повышает риск развития МДС.
- Генетические синдромы: пациенты с некоторыми наследственными синдромами имеют более высокий риск развития МДС. С развитием МДС связаны следующие генетические заболевания: анемия Фанкони, синдром Швахмана-Даймонда, анемия Даймонда-Блекфена, семейная тромбоцитопатия с предрасположенностью к развитию миелоидных злокачественных опухолей, тяжелая врожденная нейтропения и врожденный дискератоз.
- Наличие МДС в семейном анамнезе: определенная генетическая мутация, присутствующая в семейном анамнезе, может повышать риск возникновения МДС.
- Курение: канцерогенные вещества, содержащиеся в табачном дыме, всасываются в кровь при ее прохождении через легкие и распространяются по всему организму.
- Воздействие факторов окружающей среды: длительный контакт с химическими веществами, которые используются в резиновой и нефтяной промышленности (бензол и др.), может повышать риск развития МДС. Кроме того, воздействие высоких доз радиации (как, например, при аварии на ядерном реакторе) также повышает риск развития заболевания.
Каковы симптомы МДС?
- Анемия (пониженное содержание эритроцитов) может вызвать следующие симптомы:
- Усталость;
- Головокружение;
- Слабость;
- Бледность кожного покрова;
- Одышка;
- Головные боли.
- Частые или тяжелые инфекционные заболевания.
- Кровоточивость и/или появление синяков при незначительном воздействии;
- Частые или сильные носовые кровотечения;
- Кровоточивость десен.
- Повышенная температура тела;
- Боли в костях;
- Потеря веса;
- Отсутствие аппетита.
Типы МДС
Система классификации ВОЗ выделяет шесть основных типов миелодиспластических синдромов:
Читайте также: