Комплекс Эйзенменгера у ребенка. Синдром (комплекс) Тауссиг — Бинга
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Отхождение обоих магистральных сосудов, выносящих кровь из сердца не от соответствующих («своих») левого и правого желудочков, а только от одного правого — большая группа нарушений нормального развития сердца, это целый спектр аномалий, объединенных одним общим названием. Если вы читаете только эту главу, потому что у вашего ребенка именно такой диагноз, то мы советуем вам вернуться немного назад и просмотреть разделы, касающиеся изолированных больших дефектов межжелудочковой перегородки, легочной гипертензии и тетрады Фалло, потому что все, что сказано там о клиническом проявлении порока, симптомах, осложнениях и последствиях, можно в полной мере отнести и к этому пороку.
Главное нарушение формирования сердца заключается в том, что аорта, которая на 5-й неделе от момента зачатия должна «повернуться» против часовой стрелки и соединиться с левым желудочком, этого не делает, а остается «сидеть» над правым желудочком целиком. Выход крови из левого желудочка осуществляется только через большой дефект межжелудочковой перегородки и хотя потоки крови в правом желудочке, естественно, смешиваются, все же остается их частичное разделение, и артериальная кровь поступает, в основном, в аорту, которая отделена от левого желудочка мышечным валиком межжелудочковой перегородки, а венозная идет преимущественно в легкие.
Иными словами, в типичных случаях и структурное нарушение, и клинические проявления имеют черты больших дефектов межжелудочковой перегородки и — соответствующие последствия. Если же к этому добавить сужение выводного тракта правого желудочка, то картина будет напоминать тетраду Фалло.
При первых вариантах ребенок бледный, без выраженных признаков цианоза, и имеется большой сброс «слева-направо», ранние признаки сердечной недостаточности и переполнения кровью сосудов легких со всеми вытекающими отсюда последствиями, которые могут привести к развитию необратимой легочной гипертензии.
При вторых — ребенок синюшный, с выраженным цианозом и большой вероятностью одышечно-цианотических приступов.
Возможности радикального хирургического лечения сегодня существуют практически для всех вариантов порока, но нужно учитывать несколько важных деталей.
Многое зависит от того, какой у ребенка дефект межжелудочковой перегородки, где он расположен в перегородке и какого он размера. Если отверстие большое и находится прямо под устьем аорты, его закрывают, создавая тоннель из продолговатой синтетической заплаты, который соединит стенки дефекта с аортой, и таким образом аорту с левым желудочком.
Но, к сожалению, дефект (а иногда и два-три дефекта) может находится в мышечной части перегородки. Это делает операцию очень травматичной, а подчас и просто невозможной технически.
Наконец, последний вариант — дефект может располагаться под началом, устьем легочной артерии. Тогда просто соединить аорту с левым желудочком внутри сердца невозможно (так называемая ).
Выбор способа хирургической помощи, зависит от очень многих обстоятельств. Главное, что вам нужно знать — вы должны обратиться к специалистам как можно раньше, т.е. как только поставлен диагноз. Не надо ждать ухудшения — вы рискуете сделать ситуацию «запущенной». Но не надо и впадать в отчаяние. Ребенка можно и нужно лечить, и для этого есть все средства.
Об операции при «подаортальном» дефекте мы рассказывали. Она — относительно проста и ее можно делать в любом возрасте, т.е. как и при больших дефектах межжелудочковой перегородки, в зависимости от клинических проявлений порока. Но, если дефект нельзя просто превратить в выводной отдел для левого желудочка, что тогда? Если диагноз установлен достаточно рано, а давление в легочной артерии растет, то для защиты сосудов легких от необратимых повреждений нужно сузить легочную артерию, наложив манжетку, как это было описано ранее.
Делая это, мы думаем о будущем, а пока — знаем, что сосуды легких не страдают. Что нас ожидает? Через несколько месяцев или лет, а в данном случае спешить, как правило, не следует, — можно поставить вопрос о возможности радикальной операции. Ребенок подрос, набрал в весе, неплохо развит, и хотя у него может усиливаться цианоз губ, особых опасений это не вызывает — причина цианоза понятна, т.к. создана хирургически. И тут существуют две возможности. Первая и наилучшая — это удалить манжетку и закрыть дефект, создав внутри полости правого желудочка тоннель между дефектом и аортой желудочка. Этот тоннель, однако, будет более длинным, и сделать его намного труднее, чем при «подаортальном» дефекте, — из-за далекого расположения в перегородке. Иногда это технически невозможно. И тогда есть второй путь — «радикальная паллиация», когда, удалив манжетку, нужно закрыть заплатами вход в правый желудочек (т.е. трехстворчатый клапан) и сделать из обоих желудочков один, работающий на большой круг, а в предсердии произвести операцию Фонтена, как это было описано для атрезии трехстворчатого клапана. Второе — не идеальный выход, но все же — выход, когда нет другой возможности. И, надо сказать, что такой путь применяется и при других сложных пороках сердца, о которых мы скажем позже.
При аномалии Тауссиг-Бинга, или отхождении сосудов от правого желудочка и огромном дефекте межжелудочковой перегородки, расположенном под легочной артерией, тактика и стадийность хирургического лечения полностью зависит от времени первого появления ребенка в специализированном хирургическом учреждении. В любом случае, вмешательство следует делать в течение первых месяцев жизни. Не будем подробно останавливаться на существующих методиках лечения этого сложного порока. Достаточно сказать, что при раннем обращении вполне возможно выполнение операции в один этап и восстановление нормального кровообращения в обоих кругах. Но если момент упущен, то не отчаивайтесь, потому что на вооружении сегодняшней хирургии имеется много способов помочь вашему ребенку , и они зависят от типа порока и возраста ребенка. В одном из последних учебников по кардиохирургии врожденных пороков сердца насчитывается 5 вариантов хирургического лечения различных типов отхождения сосудов от правого желудочка. Мы описали только некоторые из них, наиболее типичные. В каждом конкретном случае можно и нужно выбрать наиболее логичный и безопасный метод.
Рецептов, которые были бы одними и теми же для разных детей — нет. Каждый случай индивидуален. Но главное — больные, которые еще 10 лет назад считались неоперабельными, сегодня могут рассчитывать на хирургическую помощь, которая не только продлит жизнь, но и изменит ее качество, сделает их полноценными членами общества.
Синдром Рубинштейна-Тейби
Синдром Рубинштейна-Тейби - генетически гетерогенное (по последним данным) наследственное заболевание, характеризующееся поражением центральной нервной системы, деформациями костей скелета и рядом других пороков развития. Симптомами этого состояния являются прогрессирующая умственная отсталость, низкий рост, расширение фаланг пальцев, полидактилия на ногах, разнообразные нарушения со стороны внутренних органов. Диагностика синдрома Рубинштейна-Тейби производится на основании данных настоящего статуса пациента, молекулярно-генетических анализов и других исследований. Специфического лечения данной патологии не существует, применяют симптоматическую терапию в зависимости от типа пороков и нарушений.
Общие сведения
Синдром Рубинштейна-Тейби - генетическое заболевание, которое сопровождается нарушениями интеллектуального и физического развития, а также разнообразными пороками скелета и внутренних органов. Впервые данная патология была выявлена в 1963 году и описана американскими педиатрами Дж. Рубинштейном и Г. Тейби - сами исследователи назвали заболевание «синдромом широкого первого пальца кистей и стоп, специфического лица и умственной отсталости». Дальнейшие исследования в области генетики подтвердили, что синдром Рубинштейна-Тейби является аутосомно-доминантной патологией, но подавляющее большинство случаев возникает вследствие спонтанных мутаций различного типа. Встречаемость составляет порядка 1:100 000-125 000, мальчики и девочки поражаются примерно с одинаковой частотой. Особенностью синдрома Рубинштейна-Тейби является повышенный риск развития различных онкологических заболеваний - главным образом, некоторых типов лейкозов, опухолей мозговых оболочек и нервных тканей.
Причины синдрома Рубинштейна-Тейби
Синдром Рубинштейна-Тейби обладает выраженной генетической гетерогенностью, однако различные типы этого заболевания не имеют каких-либо особенностей клинического течения. Наиболее часто причиной этой патологии выступают повреждения гена CREBBP, который расположен на 16-й хромосоме. Он кодирует так называемый CREB-связывающий белок, являющийся важным транскрипционным фактором, контролирующим экспрессию огромного количества генов. Появление в гене CREBBP различных изменений (точечных мутаций, транслокаций, микроделеций в 16-й хромосоме) приводит либо к образованию дефектного белка, неспособного выполнять свои функции, либо к полной блокировке его выделения. Это становится причиной развития синдрома Рубинштейна-Тейби.
В последние годы молекулярно-генетические исследования данной патологии подтвердили, что в ее развитии играют роль и мутации гена EP300, который располагается на 22-й хромосоме. Продуктом экспрессии данного гена является протеин p300, который, как и CREB-связывающий белок, относится к группе транскрипционных факторов. По данным исследований, идентифицировать характер генетического дефекта при синдроме Рубинштейна-Тейби удается в 55-60% случаев, примерно у половины пациентов обнаруживаются мутации гена CREBBP, еще у 40-45% выявляются делеции и другие хромосомные перестройки, затрагивающую 16-ю хромосому, и лишь у 3% больных наблюдаются мутации гена EP300. Все это указывает на возможность участия других генов в развитии этого заболевания.
Дефект белка-фактора транскрипции или его малое выделение (по причине гаплонедостаточности) приводит к многочисленным нарушениям, обусловленным недостаточной стимуляцией ряда других генов. Это можно заметить по характерной клинической картине синдрома Рубинштейна-Тейби. Оба транскрипционных фактора влияют на активность образования новых связей между нейронами, формирование долговременной памяти, стимуляцию иммунного ответа, развитие репродуктивной системы. Недостаточность этих белков ведет к целому каскаду патологических процессов в организме, что и составляет клинику синдрома Рубинштейна-Тейби. Кроме того, вышеуказанные транскрипционные факторы также влияют на активность антионкогенов, поэтому при их дефекте значительно возрастает риск развития злокачественных новообразований.
Симптомы синдрома Рубинштейна-Тейби
Симптоматика синдрома Рубинштейна-Тейби характеризуется значительным разнообразием у разных больных, что отражается на тяжести течения заболевания и его прогнозе. Обычно уже при рождении можно обнаружить некоторые признаки этой патологии - деформации лица и черепа (микро- или брахицефалия, расширение переносицы, эпикант, клювовидный нос), высокое арковидное нёбо, изменение формы и положения ушных раковин, расширенные фаланги пальцев, особенно первых. На ногах у больных синдромом Рубинштейна-Тейби нередко выявляется полидактилия. Иногда уже при рождении определяются признаки пороков развития внутренних органов - бледность или цианоз (при поражении легких или сердца), длительная желтуха новорожденных, крипторхизм.
При дальнейшем течении синдрома Рубинштейна-Тейби отмечается прогрессирующее отставание ребенка в физическом и умственном развитии от здоровых сверстников, затрудненное развитие речи и моторных навыков. Также наблюдается косоглазие, миопия. Выражение лица больных характеризуется гримасой, напоминающей улыбку, возникает гирсутизм, примерно у половины пациентов определяется наличие красного невуса в области лба, затылка или шеи. В старшем возрасте у больных синдромом Рубинштейна-Тейби выявляются искривления позвоночного столба различного характера и низкий рост (не более 150-160 сантиметров). Могут определяться деформации грудной клетки и (реже) другие скелетные аномалии, у мальчиков в большинстве случаев отмечается крипторхизм.
Постоянным признаком синдрома Рубинштейна-Тейби является умственная отсталость. Как правило, диагностируется ЗПР, олигофрения, значительная задержка речевого развития по сравнению со здоровыми сверстниками. Отмечаются нарушения концентрации внимания, больные легко отвлекаются на посторонние раздражители при выполнении какого-либо задания, возможны резкие перепады настроения. При этом лица с синдромом Рубинштейна-Тейби хорошо идут на контакт, легко социализируются. Среди других неврологических симптомов заболевания нередко обнаруживается плохая координация движений, изредка наблюдаются судорожные приступы.
Синдром Рубинштейна-Тейби может также проявляться различными нарушениями со стороны внутренних органов - головного мозга, сердца, почек и мочевыделительных путей, терминальных отделов пищеварительной системы. Кроме того, у таких больных значительно повышается риск возникновения онкологических заболеваний, в основном развивающихся в раннем возрасте. К ним относят различные формы лейкозов, меланому, некоторые типы лимфом. Поэтому до пубертатного периода больные синдромом Рубинштейна-Тейби должны периодически проходить обследования у онколога для ранней диагностики злокачественных новообразований.
Диагностика и лечение синдрома Рубинштейна-Тейби
Для выявления синдрома Рубинштейна-Тейби используют данные общего осмотра больного, рентгенологических исследований и молекулярно-генетических анализов, вспомогательную роль могут играть УЗИ и МРТ. При осмотре определяются характерные для этого заболевания аномалии развития лица (изменение формы и размеров черепа и носа, антимонголоидный разрез глаз), укорочение и расширение фаланг больших пальцев на руках и ногах, иногда - полидактилия. У взрослых больных синдромом Рубинштейна-Тейби также выявляют уменьшение роста, глубокую умственную отсталость, искривления позвоночника. На рентгенограммах можно увидеть изменения костей фаланг, позвоночника и грудной клетки, костный возраст несколько отстает от фактического.
Молекулярно-генетическая диагностика синдрома Рубинштейна-Тейби выполняется врачом-генетиком, который может использовать множество методов для определения этого заболевания. Точечные мутации в генах CREBBP и ЕР300 выявляются посредством прямого автоматического секвенирования кодирующей последовательности. Методика FISH используется в том случае, когда подозревается наличие делеций или транслокаций на 16-й хромосоме, также приводящих к развитию синдрома Рубинштейна-Тейби. Так как на основании фенотипических данных крайне сложно определить возможный тип генетического дефекта, в рамках диагностики этого заболевания нередко приходится применять сразу несколько техник современной молекулярной генетики.
Вспомогательные методы диагностики позволяют выявить сопутствующие нарушения внутренних органов при синдроме Рубинштейна-Тейби. К таким методам относят ультразвуковые исследования (УЗИ почек и мочевыделительных путей, ЭхоКГ), электрокардиографию, магнитно-резонансную томографию и другие методики. Нередко у больных синдромом Рубинштейна-Тейби диагностируют врожденные пороки сердца (открытый Боталлов проток, дефект межжелудочковой перегородки), аритмии, аномалии почек (удвоение, гипоплазия) и мочевыделительных путей (атрезии на различных участках). Также при этом заболевании нарушается формирование мозолистого тела головного мозга, что определяется при помощи магнитно-резонансной томографии. У части больных синдромом Рубинштейна-Тейби отмечаются пороки развития легких и пищеварительной системы.
Специфического лечения данной патологии на сегодняшний момент не существует, все терапевтические мероприятия сводятся к облегчению симптомов, коррекции аномалий и пороков развития, угрожающих жизни больных. Назначаются ноотропные средства, препараты кальция и витамина Д, для уменьшения выраженности умственной отсталости рекомендуют специализированную психологическую помощь. В ряде случаев при синдроме Рубинштейна-Тейби применяют хирургические методы лечения - для устранения пороков сердца, аномалий развития прямой кишки и мочевыделительных путей, крипторхизма. Также может потребоваться лечение у офтальмолога, а при признаках развития злокачественного новообразования - консультация и лечение у онколога.
Прогноз и профилактика синдрома Рубинштейна-Тейби
Согласно мнению большинства специалистов, прогноз синдрома Рубинштейна-Тейби чаще всего неопределенный или неблагоприятный. Это связано с тем, что данное заболевание характеризуется значительным спектром разнообразных нарушений - от относительно безопасных для жизни (легкие скелетные аномалии, умственная неполноценность) до ярко выраженных, способных привести к смерти в раннем возрасте (тяжелые пороки развития сердца, легких, почек). Кроме того, значительно повышен риск развития онкологических патологий. Поэтому прогноз синдрома Рубинштейна-Тейби составляется строго индивидуально, исходя из конкретной клинической картины и общего состояния больного. Каких-либо специфических методов профилактики этого заболевания из-за его частого спонтанного развития на сегодняшний день не разработано.
Болезнь Бинсвангера
Болезнь Бинсвангера - субкортикальная энцефалопатия, прогрессирующее поражение белого вещества головного мозга. В подавляющем большинстве случаев обусловлена артериальной гипертонией, реже провоцируется амилоидной ангиопатией и генетически обусловленной ангиопатией САDАSIL. Проявляется расстройствами мышления и познавательной деятельности, прогрессирующим слабоумием, нарушениями ходьбы и расстройствами функций тазовых органов. На поздних стадиях пациенты полностью беспомощны и нуждаются в постоянном уходе. Лечение - симптоматическая и патогенетическая терапия основного заболевания, коррекция неврологических и психических нарушений.
Болезнь Бинсвангера - прогрессирующая субкортикальная энцефалопатия. Обусловлена сосудистыми нарушениями в результате стойкого повышения артериального давления. Впервые была описана в 1894 году немецким невропатологом и психиатром Отто Бинсвангером. Стала широко известной, благодаря ученику Бинсвангера Альцгеймеру. В течение долгого времени не признавалась большинством невропатологов и психиатров из-за недостаточно четкого описания и малого количества исследованных случаев заболевания.
Отношение к болезни Бинсвангера изменилось после появления МРТ и КТ головного мозга. Данные, полученные при проведении компьютерной томографии и магнитно-резонансной томографии, подтвердили общий характер изменений белого вещества головного мозга у больных артериальной гипертонией с сопутствующей прогрессирующей деменцией. В настоящее время заболевание считается достаточно широко распространенным, исследованиям болезни Бинсвангера посвящено множество публикаций. Лечение болезни Бинсвангера осуществляют невропатологи и специалисты в области психиатрии.
Этиология и патогенез болезни Бинсвангера
Основной причиной развития данного заболевания является стойкое повышение артериального давления при эссенциальной или симптоматической гипертонии. Специалисты отмечают, что у пожилых людей болезнь может провоцироваться постоянным снижением или резкими суточными колебаниями артериального давления. В литературе встречаются упоминания о пациентах, у которых подкорковая энцефалопатия возникла на фоне нормального артериального давления. В отдельных случаях пусковым фактором становятся другие заболевания, сопровождающиеся поражением сосудов головного мозга (амилоидная ангиопатия и церебральная аутосомно-доминантная ангиопатия).
По различным данным, больные с артериальной гипертензией составляют от 75 до 98% от общего количества пациентов с болезнью Бинсвангера. При симптоматической и эссенциальной гипертонии первые признаки подкорковой энцефалопатии обычно обнаруживаются в возрасте 50 лет и старше, средний возраст больных составляет 54-66 лет. У пациентов, страдающих генетически обусловленной ангиопатией, симптомы прогрессирующей деменции появляются в среднем или молодом возрасте. Амилоидная ангиопатия обычно диагностируется у лиц преклонного возраста.
Патологические изменения белого вещества обусловлены склерозом мелких артерий головного мозга, сопровождающимся уменьшением просвета сосудов и увеличением толщины сосудистой стенки. Тотальное ухудшение кровоснабжения подкорковых областей вызывает диффузные изменения белого вещества атрофического характера. В белом веществе появляются множественные кровоизлияния, кисты и мелкие очаги разрушения, его плотность снижается, оно уменьшается в объеме и частично замещается жидкостью (гидроцефалия). Желудочки мозга расширяются.
Симптомы болезни Бинсвангера
Для сосудистой подкорковой энцефалопатии характерны нарушения походки, расстройства функции тазовых органов, нерезко или умеренно выраженные неврологические симптомы (обычно проходящие), прогрессирующее слабоумие, волевые и эмоциональные нарушения. Болезнь развивается постепенно. Возможно как непрерывное прогредиентное течение, так и медленное прогрессирование с длительными периодами стабилизации. Усугубление симптоматики обычно напрямую связано с продолжительным повышением артериального давления. Исходом становится полная беспомощность, неспособность к самообслуживанию и отсутствие контроля над функциями тазовых органов.
Слабоумие имеет характер, типичный для сосудистых заболеваний головного мозга. Наблюдается ухудшение памяти, снижение уровня суждений, замедление и неустойчивость психических процессов. Часто выявляется «эмоциональное недержание» - выраженная неспособность сдерживать эмоции с преобладанием астенических реакций. Возможны продолжительные периоды стабилизации и даже временный регресс имеющихся нарушений.
В зависимости от преобладающей симптоматики выделяют дисмнестическое, амнестическое и псевдопаралитическое слабоумие. При дисмнестическом слабоумии наблюдается нерезко выраженное снижение памяти и интеллекта, замедление физических и психических реакций. Критика к собственному состоянию и поведению незначительно нарушена. В клинической картине амнестического слабоумия превалируют выраженные нарушения памяти на текущие события при сохранении прошлых воспоминаний. Для псевдопаралитического слабоумия характерны устойчивое, однообразное благодушное настроение, незначительные расстройства памяти и выраженное снижение критики.
Клиническая картина при всех формах слабоумия весьма вариативна, может выявляться преобладание как корковых, так и подкорковых нарушений, при этом корковые нарушения сопровождаются более заметным снижением интеллектуально-мнестической деятельности. В отдельных случаях наблюдаются эпилептические припадки. Слабоумие сопровождается нарушениями в эмоциональной и волевой сфере. Возможны неврозоподобные явления, повышенная истощаемость и снижение настроения. На поздних стадиях болезни отмечается ограничение интересов, эмоциональное оскудение и потеря спонтанности.
Нарушения походки, как и слабоумие, прогрессируют постепенно. Вначале шаги становятся более мелкими, пациент начинает шаркать ногами, тяжело отрывает ноги от земли. В последующем нарушается автоматизм ходьбы, походка становится медленной и осторожной, все движения контролируются сознательно, как будто больной идет по скользкому льду. Выделяют следующие признаки нарушений походки при болезни Бинсвангера: уменьшение длины шага, замедление ходьбы, потребность в повышенной устойчивости, трудности при начале ходьбы и снижение устойчивости при поворотах.
Нарушения функций тазовых органов на начальных стадиях проявляются гиперактивностью мочевого пузыря - внезапными сильными позывами на мочеиспускание и учащением мочеиспускания, которые при прогрессировании болезни трансформируются в императивное недержание мочи. Наряду с нарушениями функция тазовых органов могут выявляться и другие неврологические симптомы, в том числе - псевдобульбарный синдром, преходящие парезы и паркинсоноподобные расстройства.
Диагностика болезни Бинсвангера
Диагноз выставляется на основании анамнеза, клинической картины, данных КТ и МРТ головного мозга. Основанием для постановки диагноза является клинически значимая деменция, подтвержденная результатами нейропсихологического исследования, в сочетании с минимум двумя признаками, характерными для болезни Бинсвангера. В числе этих признаков - наличие заболевания, сопровождающегося системным поражением сосудов (симптоматическая или эссенциальная гипертония, аритмия, инфаркт миокарда, сахарный диабет и т. д.); неврологические нарушения (расстройства ходьбы, спастический мочевой пузырь, императивные позывы на мочеиспускание или императивное недержание мочи); свидетельства сосудистой патологии головного мозга (инсульт в анамнезе, очаговые неврологические симптомы).
В некоторых случаях болезнь Бинсвангера требуется дифференцировать с болезнью Альцгеймера. В процессе дифференциальной диагностики используют ишемическую шкалу Хачинского, учитывающую постепенность или внезапное начало болезни, прогредиентность или ступенеобразность течения, выраженность нарушений личности, а также наличие или отсутствие депрессивных расстройств, эмоциональной неустойчивости, артериальной гипертонии, атеросклероза, инсультов и неврологических расстройств.
Лечение болезни Бинсвангера
Лечение осуществляют неврологи и психиатры в сотрудничестве с терапевтами, кардиологами, эндокринологами и другими специалистами (в зависимости от основной патологии). План лечения составляют индивидуально, с учетом стадии и особенностей течения основного заболевания, выраженности неврологических, мнестических, эмоциональных и интеллектуальных нарушений. Проводят этиопатогенетическую и симптоматическую терапию, назначают лекарственные средства для коррекции эмоционального состояния и улучшения когнитивных функций. В основе болезни лежит повышение АД, поэтому важной задачей терапии становится его нормализация. При этом необходимо не допускать чрезмерного снижения артериального давления, постоянно поддерживая показатели в пределах 120/80 мм рт. ст., поскольку гипотония может способствовать усугублению возникших нарушений.
Для профилактики нарушений мозгового кровообращения назначают антитромбоцитарные средства. При наличии сопутствующих болезней сердца используют антикоагулянты. Для улучшения когнитивных функций применяют ингибиторы МАО, ноотропы, антиоксиданты, нейротрофические, мембраностабилизирующие и антихолинэстеразные препараты. При депрессивных расстройствах осуществляют терапию с использованием антидепрессантов (преимущественно - ингибиторов обратного захвата серотонина).
Комплекс Эйзенменгера. Лечение в клинике Шнайдер
Комплекс Эйзенменгера — это комбинированный врожденный порок сердца у детей, при котором существуют несколько аномалий развития:
— обширного, высоко расположенного дефекта перепончатой части перегородки между желудочками;
— расположенной справа аорты, в результате чего кровь в этот магистральный сосуд поступает как из левого, так и из правого желудочка сердца;
— гипертрофии мышечного слоя правого желудочка сердца.
Лечение порока сердца у детей в детской кардиологии клиники Шнайдер
Достаточно часто к указанным врождённым аномалиям сердечно-сосудистой системы присоединяются дефекты перегородки между предсердиями, незаращение аортального протока, коарктация аорты.
Следует различать комплекс Эйзенменгера и одноименный синдром. Если комплекс Эйзенменгера предполагает наличие трёх указанных дефектов, то синдром Эйзенменгера обозначает целый комплекс симптомов, который формируется вследствие венозно-артериального шунтирования крови. Специалисты отделения детской кардиологии клиники Шнайдер занимаются диагностикой и лечением детей в Израиле на высшем уровне.
Комплекс Эйзенменгера. Симптомы порока сердца у детей
Довольно часто болезнь не дает ярких клинических симптомов. Цианоз обычно развиваются после десяти лет, в пубертатном периоде или в после завершения полового созревания. На протяжении достаточно длительного времени цианоз может быть выражен умеренно, регистрироваться только при интенсивной физической нагрузке. При этом доктора клиники Шнайдер отмечают, что появление цианоза при данном пороке сердца считается плохим признаком и требует немедленного лечения.
К наиболее частым жалобам при комплексе Эйзенменгера следует отнести головную боль, повышенную утомляемость, частые обмороки и выраженную одышку. Характерно, что облегчение состояния наблюдается в положении сидя на корточках. Часто регистрируются инфекционные процессы дыхательных путей, а также носовые кровотечения и кровохарканье.
При обследовании в детской кардиологии клиники Шнайдер опытные врачи проводят наиболее информативные инструментальные методы исследования: ангиокардиографию, кардиометрию, зондирование камер сердца.
Эффективное лечение комплекса Эйзенменгера у детей в Израиле
Лечение комплекса Эйзенменгера у детей в Израиле в клинике Шнайдер проводят только опытные специалисты. Самым эффективным способом лечения считается хирургическая операция. Оперативное закрытие имеющегося дефекта в межжелудочковой перегородке до появления первых явлений цианоза позволяет обеспечить высокое качество жизни и существенно увеличить её продолжительность.
В отделении детской кардиологии клиники Шнайдер есть вся необходимая сложная аппаратура, с которой работают выдающиеся врачи, владеющие техникой устранения сложных врождённых пороков. Одновременно с закрытием дефекта необходимо устранить аномальное расположение ствола легочной артерии. Существенно осложняет ход операции необходимость имплантирования искусственного аортального клапана.
Специалисты клиники Шнайдер проводят эффективные операции даже при раннем появлении цианоза. Таким образом неблагоприятный прогностический признак не мешает врачам успешно проводить лечение десяткам пациентов.
ТАУССИГ-БИНГА СИНДРОМ
ТАУССИГ-БИНГА СИНДРОМ (Н. В. Taussig, род. в 1898 г., амер. кардиолог; R. J. Bing, род. в 1909 г., амер. врач; син.: транспозиция аорты и левопозиция легочного ствола, неполная транспозиция магистральных сосудов с отхождением легочного ствола от двух желудочков) — врожденный порок сердца, при к-ром аорта отходит от правого желудочка, а легочный ствол от обоих желудочков при высоко расположенном дефекте межжелудочковой перегородки.
Формирование Т.— Б. с. связано с тем, что в процессе эмбрионального развития артериального конуса не происходит сдвига луковицы сердца к средней линии. В результате недостаточного развития луковично-желудочкового выступа одновременно образуется дефект межжелудочковой перегородки.
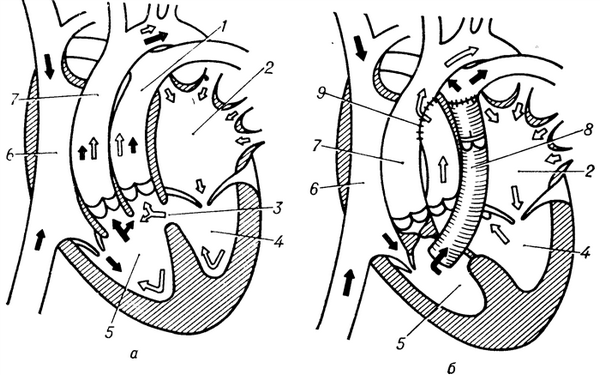
Схематическое изображение сердца и магистральных сосудов на фронтальном разрезе при синдроме Тауссиг— Бинга до (а) и после (б) радикальной коррекции порока (по методу Смита): 1 — легочный ствол; 2 — левое предсердие; 3 — дефект межжелудочковой перегородки; 4 — левый желудочек; 5 — правый желудочек; 6 — правое предсердие; 7 — аорта; 8 — клапансодержащий протез легочного ствола; 9 - анастомоз резецированного отрезка легочного ствола и аорты конец в бок; направление движения венозной крови показано темными стрелками, артериальной крови — светлыми стрелками.
Основные проявления порока определяются особенностями создающейся при этом гемодинамики: кровь из правого желудочка поступает в аорту, а также в легочный ствол (рис., а). Давление в правом желудочке и аорте одинаковое, а в левом желудочке давление равно или немного меньше, чем в правом. Характерной особенностью Т.— Б. с. является стойкая легочная гипертензия (см. Гипертензия малого круга кровообращения) при отсутствии клапанного стеноза легочного ствола, приводящая к склерозу сосудов малого круга кровообращения и уменьшению эффективности легочного кровотока. Насыщение крови кислородом в аорте при Т.— Б. с. ниже, чем в легочном стволе, что приводит к гипоксемии различной степени. Диаметр легочного ствола при Т. — Б. с. в 2—3 раза больше диаметра аорты.
Дети с Т.— Б. с. заметно отстают в развитии. У больных отмечается цианоз с рождения, деформация дистальных (ногтевых) фаланг в виде барабанных палочек, выраженная одышка, однако без одышечно-цианотических приступов. Границы сердца перкуторно умеренно увеличены; отмечается систолическое дрожание во втором межреберье слева от грудины и несколько ниже. Аускультативно определяются акцент II тона и пансистолический шум. Рентгенологически чаще всего сердце нормальных размеров, легочный рисунок по периферии обеднен, корни легких расширены. Слева резко выбухает дуга легочного ствола, пульсация к-рого усилена. На ЭКГ — отклонение электрической оси вправо, гипертрофия правого желудочка.
Диагноз Т. —Б. с. подтверждается данными катетеризации полостей сердца (см. Катетеризация сердца) и ангиокардиографии (см.), позволяющими выявить высокое давление в правом желудочке и стволе легочной артерии, высокое насыщение кислородом крови в ней. При селективной ангиографии в двух проекциях определяется раннее и интенсивное контрастирование аорты, при правой вентрикулографии — одновременное, но более слабое контрастирование легочного ствола; расположение аорты кпереди от легочного ствола, более широкий диаметр легочного ствола по сравнению с аортой.
Лечение — оперативное вмешательство. Операции в основном паллиативные — в раннем возрасте сужение легочного ствола по Мюллеру, создание искусственного дефекта межпредсердной перегородки для артериализации крови правых отделов сердца; соединение нижней полой вены с левым предсердием, а правых легочных вен — с правым предсердием (операция Баффеса). Паллиативные методы однако не устраняют легочную гипертензию, к-рая в конечном итоге приводит к гибели больного.
Смит (E. Е. Smith) с соавт. в 1982 г. предложил метод радикальной коррекции с помощью клапансодержащего протеза легочного ствола (рис., б). Операция заключается в создании адекватных путей оттока крови от правого желудочка. Для этого легочный ствол отсекают у бифуркации и анастомозируют с аортой конец в бок, устье аорты ушивают; кровоток из правого желудочка направляют в малый круг кровообращения через клапансодержащий протез, анастомозируя его с дистальным отрезком легочного ствола. Эта оригинальная и сложная операция признается перспективной, разрабатываются новые ее методики. Материалов по исходу и отдаленным результатам этой операции, разумеется, пока нет.
Прогноз при Т.— Б. с. неблагоприятный. Продолжительность жизни зависит от тяжести анатомических изменений, выраженности легочной гипертензии и общей гипоксии. Основная причина смерти — правожелудочковая недостаточность.
Библиография: Банки Г. Врожденные пороки сердца и крупных сосудов, пер. с англ., с. 87, М., 1980; Константинов Б. А. Аномалия Тауссиг — Бингл, в кн.: Частн. хир. болезней сердца и сосудов, под ред. В И. Бураковского и С. А. Колесникова, с. 230, 'М., 1967; В a f f e s T. G. A new method for surgical correction of transposition of aorta and pulmonary artery, Surg. Gynec. Obstet., v. 102, p. 227, 1956; Go or D. A. a. Lillehei C. W, Congenital malformations of heart, p. 203, N. Y. a. o., 1975; Smith E. E. a. o. A new technique for correction of the Taussig — Bing anomaly, J, thorac. cardiovasc. Surg., v. 83, p. 901, 1982; Taussig H. B. a. Bing R. J. Complete transposition of aorta and levoposition of the pulmonary artery, Amer. Heart J., v. 37, p. 551, 1949; Van Praagh R. What is the Taussig — Bing malformation? Circulation, v. 38, p. 445, 1968.
Читайте также: